The Phakomatoses
The term phakomatosis was first coined by ophthalmologist Jan van der Hoeve in 1920 to compare similarities between neurofibromatosis and tuberous sclerosis.1 In doing so, he defined the term phakoma to describe congenital tumors in multiple tissues distinct from nevi by the lack of nevus cells.2 When he coined the term, strict inclusion or exclusion criteria for this category were omitted. Over time, because of this looseness of definition, many conditions have been included in the category. Another popular term for these conditions is neurocutaneous disorders.
The syndromes that are covered in this chapter are the more common phakomatoses, as well as the conditions that are the most relevant to neurosurgeons. They include tuberous sclerosis, Sturge-Weber syndrome, von Hippel-Lindau sydrome, hereditary hemorrhagic telangiectasia, basal cell nevus syndrome, and neurocutaneous melanosis. Neurofibromatosis is discussed in a separate chapter but is a member of this group. Other syndromes that could be included in this category are ataxia telangiectasia, incontinentia pigmenti, and Wyburn-Mason syndrome.
49.1 Tuberous Sclerosis Complex
49.1.1 Introduction
Tuberous sclerosis complex (TSC) is a genetic disorder with variable penetrance that causes multiple-organ dysfunction. Tumors can form in the brain, kidneys, heart, eyes, lungs, and skin. In 1862, the constellation of findings was first noted by von Reckinghausen.3 However, the disorder did not receive its name until Magloire Bourneville applied the term sclérose tubéreuse to describe the gross appearance of the autopsy findings.4 The disorder is relevant to neurosurgeons because 85% of children have neurologic sequelae, including epilepsy, tumors, and behavioral and psychological problems.5
49.1.2 Epidemiology
From 1 in 6,000 to 1 in 14,000 children younger than age 10 are estimated to have TSC,6,7 but the prevalence may be higher because of the variable severity of disease expression.8 When the difference in expression is accounted for, the birth incidence is estimated to be 1 in 5,800.9 There are no known demographic categories that are predominantly affected.
49.1.3 Clinical Presentation
The most common cutaneous finding is hypopigmented macules, also known as ash leaf spots, which are present in 90 to 98% of patients with TSC patients versus only 4.7% of the general population (▶ Fig. 49.1).5,10,11 These lesions are most easily detected with a Woods lamp and are often located on the trunk or buttocks.11 Bilateral facial angiofibromas (also known as adenoma sebaceum), which form a butterfly pattern over the bridge of the nose and malar eminences, are present in 80% of children with TSC who are older than 5 years (▶ Fig. 49.2).11 Shagreen patches, present in 54% of patients older than 5 years, are connective tissue nevi found commonly on the lumbosacral flanks or dispersed over the trunk and thighs.12 Forehead fibrous plaques are another type of angiofibroma found in about 36% and may be present at birth.12 Periungual fibromas (also known as Koenen tumors), which usually do not develop until age 15 to 29, are more common in women and preferentially form on toes. Molluscum fibrosum pendulum (also known as a skin tag) is commonly found in adults and is located on the neck, groin, axillae, and flexor surfaces of limbs.11 Dental pits are present in 90% of patients with TSC, whereas they are present in only 9% of the general population.13
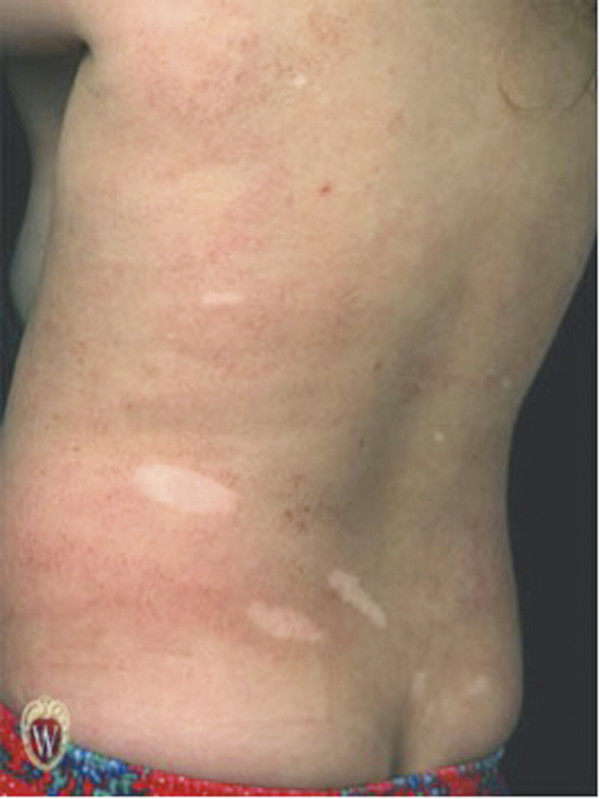
Fig. 49.1 An 8-year-old boy with hypopigmented macules (also known as ash leaf spots). These lesions are most often located on the trunk or buttocks.
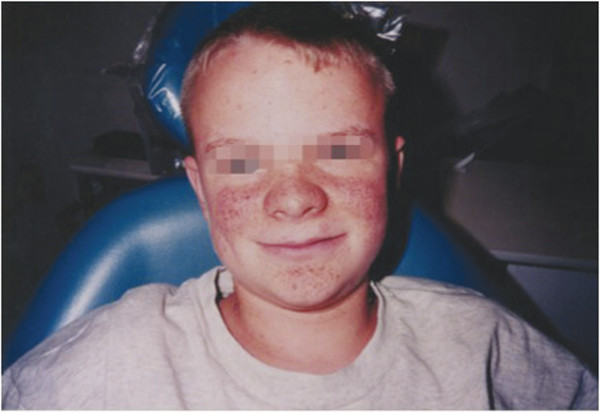
Fig. 49.2 An 11-year-old boy with bilateral facial angiofibromas (also known as adenoma sebaceum), which form a butterfly pattern over the bridge of his nose and malar eminences.
Cardiac rhabdomyomas, an almost exclusively pediatric manifestation of TSC, are a primary finding in fetuses and infants. Of infants with cardiac rhabdomyomas, 96% are diagnosed with TSC.14 Generally, these lesions are asymptomatic and detected on fetal ultrasound or when a murmur is present. Occasionally, they can be large enough to cause cardiac output dysfunction or significant arrhythmias. These tumors regress throughout early childhood, with the most dramatic reduction occurring during the first 3 years of life.15,16
Kidney complications are the most common tuberous sclerosis–related cause of death.17 The renal manifestations include renal angiomyolipomas, renal cysts, and renal cell carcinoma (RCC). Although renal angiomyolipomas are predominantly present in adults, up to 16% of children younger than 2 years old can be affected and may require treatment, either through embolization or surgical resection.18,19 Renal cysts may be present but are usually asymptomatic. RCC is present in 2 to 3% of patients with TSC and is usually diagnosed during childhood.20
Although extremely rare in pediatric TSC, pulmonary lymphangiomyomatosis is a progressive disease process that begins with shortness of breath, cough, and chest pain. It can be very difficult to treat.21
On ophthalmologic examination, retinal hamartomas are found in 40 to 50% of patients. These hamartomas have variable morphological appearances but rarely affect vision.22
Neurologic sequelae of TSC include epilepsy, neoplasms, hydrocephalus, and neurocognitive or behavioral dysfunction. On magnetic resonance (MR) imaging, affected patients typically exhibit multiple characteristic structural abnormalities, including cortical tubers, which may or may not be calcified; subependymal nodules, which do not enhance; and subependymal giant cell astrocytomas (SEGAs), which are enhancing, noncalcified lesions near the foramen of Monro (▶ Fig. 49.3). These lesions contribute to the neurologic manifestations listed above.7 The tubers can be located anywhere in the cortex; however, they are most commonly located in the frontal and parietal lobes. They represent focal areas of cortical dysplasia and appear as expanded gyri with high intensity on all MR imaging sequences (▶ Fig. 49.4). Tubers do not generally enhance with contrast. Subependymal nodules have the appearance of periventricular “candle drippings” that do not enhance.23
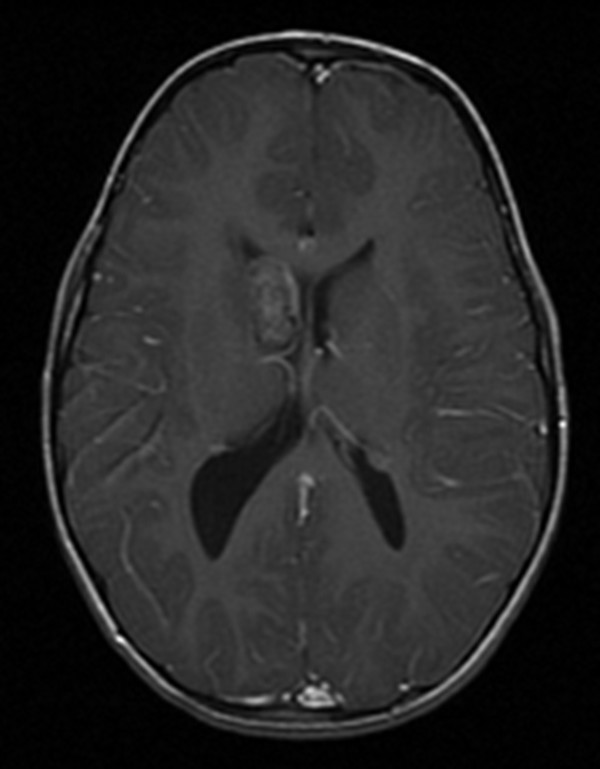
Fig. 49.3 Magnetic resonance imaging of the brain of a 4-year-old girl with tuberous sclerosis complex. This axial T1-with-contrast sequence demonstrates a subependymal giant cell astrocytoma, which is an enhancing, noncalcified lesion near the foramen of Monro.
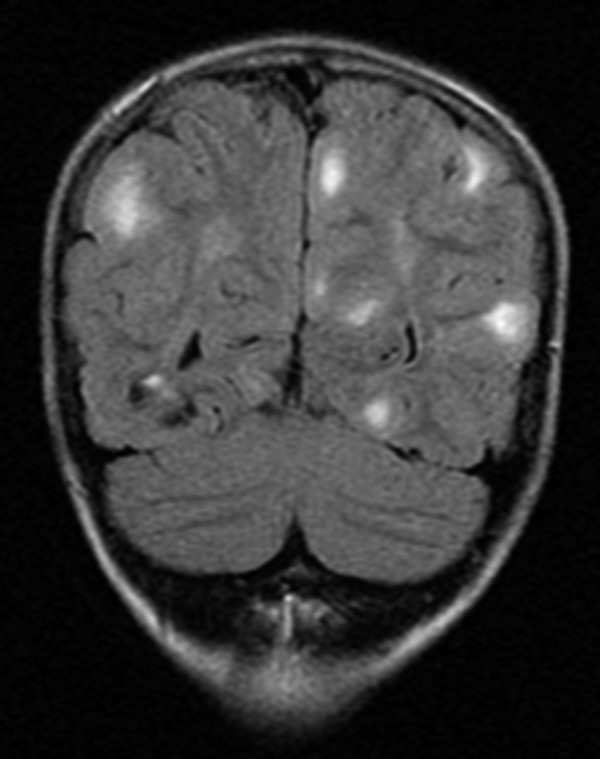
Fig. 49.4 Magnetic resonance (MR) imaging of the brain of a 4-year-old girl with tuberous sclerosis complex. This coronal fluid-attenuated inversion recovery (FLAIR) sequence demonstrates tubers, which represent focal areas of cortical dysplasia and appear as expanded gyri with high intensity on all MR imaging sequences. Tubers do not generally enhance with contrast.
Epilepsy is associated with the cortical tubers; however, it is not clear whether the tubers themselves or the perituberal cortex is epileptogenic.24,25 The prevalence of epilepsy in TSC is reported to be 80 to 90%.9 In children who develop epilepsy, seizures typically begin in the first year of life, and 85% of cases will be refractory to medical treatment. Usually, the seizures are focal at first; however, they can be preceded by or progress to infantile spasms. Infantile spasms usually evolve into other types of seizures, oftentimes leaving children with multiple seizure types.26 These children are at an increased risk for developing neurocognitive disabilities.27 Generally, children with TSC2 genetic mutations are at higher risk for developing more severe seizure phenotypes.28
SEGAs are tumors near the midline in close proximity to the foramen of Monro that develop in 5 to 15% of patients with TSC.29,30 They most commonly occur in the first two decades of life; the mean age at presentation is 11 years.31 SEGAs grow slowly and are generally 2 to 3 cm at the time of diagnosis. As World Health Organization (WHO) grade I tumors, they generally cause symptoms from mass effect, either through hydrocephalus or direct compression of the deep nuclei.32 On imaging, they can be difficult to differentiate from subependymal nodules. However, if a lesion is larger than 12 mm, enhances, and is near the foramen of Monro, it is likely an SEGA.33
Neurocognitive function varies from nearly normal to severely disabled, the latter often reflecting autistic or other neurobehavioral disorders. About 30% of children are extremely impaired, without the ability to live an independent life. However, 50% may have a normal IQ but still have some cognitive deficits.34 There are high rates of autism associated with TSC.35,36 Poor cognitive outcomes have been linked to intractable seizures, TSC2 mutation, and the location of cortical tubers.37
49.1.4 Pathology and Genetics
TSC is an autosomal-dominant inherited disorder. However, nearly two-thirds of cases are believed to result from sporadic mutations.9,38 The mutations have been identified as affecting two tumor suppressor genes, tuberous sclerosis complex 1 (TSC1), and tuberous sclerosis complex 2 (TSC2), and have been mapped to chromosomes 9q34 and 16p13.3, respectively.39,40 Of the sporadic mutations, TSC2 mutations are the most common, responsible for 75 to 80% of cases.41 TSC1 and TSC2 encode the proteins hamartin and tuberin, respectively. Under normal conditions, the proteins act as a dimer that activates a guanosine triphosphatase (GTPase). This acts as a negative downstream regulator of the mammalian target of rapamycin (mTOR), which has been the therapeutic target of rapamycin and similar drugs.42 Dysregulated mTOR activation has been implicated in the development of SEGAs and of cortical dysgenesis.43 Patients with the same genotype, even within a family, can exhibit very different phenotypes. Although there are exceptions, generally TSC2 mutations have more severe clinical manifestations than TSC1 mutations.41,44
49.1.5 Diagnostic Studies
A consensus conference defined criteria for the diagnosis of TSC. These criteria include major and minor features, and the diagnosis is made when a patient exhibits two major or one major and two minor features. The features include clinical signs and radiologic findings that are specific for tuberous sclerosis.45 These diagnostic criteria do not include genetic testing. Genetic testing is informative if the results are positive; however, it detects only 85 to 90% of TSC mutations, which is considered a relatively low sensitivity.46
When a child is diagnosed with TSC, many diagnostic studies are needed to assess the degree of systemic involvement (see box “▶ Tuberous Sclerosis Management”).7 Throughout childhood, the patient undergoes surveillance monitoring to identify the frequent complications of TSC that are treatable if diagnosed early but are highly likely to cause morbidity or mortality if the diagnosis is delayed (see box “▶ Tuberous Sclerosis Management”). Therefore, testing is directed toward diagnosing renal angiomyolipomas, SEGAs, cardiac rhabdomyomas, and pulmonary lymphangiomyolipomas, because these are the major causes of premature mortality in TSC.7,17 Any of these evaluations should be repeated more frequently when clinically indicated.7
Tuberous Sclerosis Management
Initial evaluation
Magnetic resonance (MR) imaging of the brain with and without contrast
Electroencephalography (EEG; less essential in older children who are developmentally normal and without seizures at the time of diagnosis)
Neurodevelopmental testing
Electrocardiography to evaluate for arrhythmias
Echocardiography if there are arrhythmias or cardiac dysfunction
Renal ultrasound
Ophthalmologic examination
Dermatologic examination for concerning cutaneous manifestations
Surveillance
MR imaging of the brain every 1 to 3 years
Renal ultrasound every 1 to 3 years
Neurodevelopmental testing repeated at school entry
Repeated electrocardiography, echocardiography, EEG, ophthalmologic evaluation, and dermatologic evaluation as clinically indicated
Data from Curatolo P, Bombardieri R, Jozwiak S. Tuberous sclerosis. Lancet 2008;372(9639):657–668.7
49.1.6 Treatment
A multidisciplinary approach is required to manage patients with TSC. The treatment of the nonneurosurgical features of TSC is beyond the scope of this chapter, but patients should undergo the previously described surveillance testing and be treated as needed by the appropriate medical specialists.7
Traditionally, surgery has been the treatment of choice for SEGAs, and it is curative with complete resection.33 These tumors are usually resected through a transcallosal or transcortical approach; however, authors have described purely endoscopic approaches.47 When complete resection cannot be achieved, patients typically experience slow growth of the tumor residual that requires further treatment.48 Gamma Knife has been used as both primary and adjuvant treatment; however, the results have not been consistently promising.49,50 Recent breakthroughs in pharmacologic therapy for SEGAs have taken advantage of mTOR pathway inhibition and have used rapamycin (sirolimus), its prodrug CCI-779 (temsirolimus), or its analogue RAD001 (everolimus) to counteract the uncontrolled mTOR pathway.31 These drugs are still currently being investigated, but it is notable that significant reduction in tumor size occurred in the majority of patients; however, the reduction was not durable after the cessation of treatment.51–53 The exact role of mTOR inhibitors is still being established through ongoing clinical trials. It is likely that there are clinical scenarios that will be managed by using mTOR inhibitors as either neoadjuvant therapy or for residual or surgically inaccessible lesions.31
Despite the use of multiple antiepileptic drugs (AEDs), many children develop intractable epilepsy.26 These patients are candidates for surgical evaluation for resection of the primary epilepsy focus or, in some cases, insertion of a vagal nerve stimulator. Another option is a ketogenic diet.54 For the treatment of infantile spasms, vigabatrin is the first-line therapy, even though there can be significant ophthalmologic complications.55 Second-line therapy includes corticosteroids or other AEDs. For infantile spasms, surgery can be an option if the infantile spasms are believed to be generated by a focal lesion.56 For focal seizures, vigabatrin is recommended if the child is younger than 1 year; other AEDs are used if the patient is older than 1 year. Surgery should be considered early as a second-line therapy if the seizures are refractory to AEDs and the patient has a surgically resectable localized lesion.56 Third-line therapy would be vagal nerve stimulation or a ketogenic diet.54,56,57
Patients for whom surgery is being considered should proceed through the usual epilepsy surgical evaluation, which may differ slightly among centers but includes noninvasive video electroencephalography (EEG), MR imaging, positron emission tomography (PET), single photon emission computed tomography (SPECT), magnetoencephalography (MEG), and neurocognitive testing. In many cases, the evaluation leads to invasive monitoring with subdural and depth electrodes.56 Sometimes, a multistage operation is required for a patient with multiple epileptogenic foci.58
Despite these epilepsy treatment options, up to one-third of patients remain resistant to treatment. Current investigations are examining the role of mTOR inhibitors for epilepsy, in parallel with the use of these agents for the treatment of SEGAs.43 In studies of mTOR inhibitors for the treatment of SEGAs, patients were noted to have a reduction in seizure frequency.53 The potential of using mTOR inhibitors to treat epilepsy is offset by the probable need for long-term therapy and potential side effects or toxicity profile, as well as the complexity of enzyme upregulation that occurs with many AEDs.
49.1.7 Prognosis
Early death in patients with TSC is most often caused by complications of renal disease.17 Early death can also be due to status epilepticus or hydrocephalus secondary to SEGAs.30
49.2 Sturge-Weber Syndrome
49.2.1 Introduction
Sturge-Weber syndrome (SWS), also known as encephalotrigeminal angiomatosis, is a sporadic congenital disorder affecting the brain, face, and eyes.59 In 1879, William Sturge first described the clinical syndrome,60 and in 1922, Frederick Weber described the radiographic findings as well as the pathophysiologic vascular steal phenomenon.61 Unlike most other phakomatoses, SWS is not inherited and is not associated with neoplasms; however, SWS is relevant to neurosurgeons because of the prevalence of intractable epilepsy and the potential need for epilepsy surgery.62
The syndrome consists of multiple clinical and radiologic findings, including a facial port-wine stain (PWS), leptomeningeal angiomatosis, angiomas of the choroid plexus, and congenital glaucoma. Three variants of SWS have been described, spanning a spectrum of disease severity. Type 1 is the traditionally described SWS, with the facial PWS, leptomeningeal angiomatosis, choroid angioma, and congenital glaucoma. Type 2 is the PWS alone without intracranial involvement. Lastly, type 3 is exclusively leptomeningeal angiomatosis and occurs in about 10% of cases.63,64
49.2.2 Epidemiology
The prevalence is estimated to be 1 in 50,000 live births. No gender or racial predilection exists.65
49.2.3 Clinical Presentation
The clinical manifestations vary in severity among patients and can progress over time. Typically at birth, patients with SWS exhibit a PWS, which is a facial capillary malformation.59 This facial stigma can have variable appearances, including that of a salmon patch; however, throughout this chapter, it is referred to as a PWS. Most often, a PWS is located in the V1 distribution of the trigeminal nerve, but it can extend into the V2 or less commonly the V3 distribution.59,66,67 In up to 10% of patients, the PWS can be bilateral (▶ Fig. 49.5).68

Fig. 49.5 An infant with a port-wine stain in a bilateral cranial nerve V1 distribution. Bilaterality is uncommon but can be present in up to 10% of patients.
Leptomeningeal angiomatosis develops in only 8 to 20% of children born with a PWS. The presence of leptomeningeal angiomatosis is more likely when the PWS is in the V1 distribution, large, or bilateral.66–68 If the PWS is unilateral, then any leptomeningeal angiomatosis is generally ipsilateral. The parietal and occipital lobes are the most common location for leptomeningeal angiomatosis, but it can cover an entire hemisphere or be bilateral.69
Epilepsy, developmental delay, cognitive deficits, headaches, spastic hemiparesis, and cerebral ischemia with transient ischemic attacks and strokes can occur.70–73 Seizures occur in nearly 75% of patients with unilateral disease and in 95% of those with bilateral disease.69,71 They begin at a median age of 6 months; among 75% of patients with SWS in whom epilepsy develops, seizures begin in the first year of life.72,73 Intractable epilepsy is associated with seizures beginning in the first year of life, but much of the natural history of this condition is still unknown.74,75 Initially, the seizure type is usually simple partial; however, some patients have complex partial seizures,75 and the seizures may cluster, alternating with periods of seizure freedom.10,75,76 Rarely, patients with SWS can have infantile spasms.77,78 In patients with isolated leptomeningeal involvement (type 3), the seizures usually begin later and are more easily controlled with AEDs.63
“Strokelike episodes” can occur, with prolonged hemiparesis persisting days to months or even permanently; these episodes are of longer duration than the usual Todd paralysis.75 Seizures, minor traumas, and migraine headaches have been reported as precipitating events.75,79,80 Progressively, patients can develop spastic hemiparesis and visual field deficits, but the frequency of these developments has not been reported.75
Headaches are a common complaint in patients who have SWS, with a frequency much higher than that in the general population.81,82 Migraines can be temporally related to seizures and strokelike episodes.80
Cognitively and psychologically, patients range from normal to severely disabled. More severe cognitive deficits have been associated with the early onset of seizures, intractable epilepsy, multiple seizure types, a greater degree of cerebral atrophy, and bilateral involvement.69,73,75 School-age children are more likely to be diagnosed with attention deficit disorders.
Glaucoma develops in 30 to 70% of patients. Of those in whom glaucoma develops, 60% present in infancy and 40% in childhood or early adulthood.73 Glaucoma is the result of ocular capillary venous vascular malformations.68
49.2.4 Pathology and Genetics
Although there is no genetic inheritance of SWS, somatic mosaicism may play a role.75 Embryologically, the constellation of findings associated with SWS point to the sixth week of fetal development for the timing of the dysfunction. In normal development, a vascular plexus forms around the cephalic portion of the neural tube and in proximity to the ectoderm that will form the facial skin. Normally, this vascular plexus regresses during the ninth week of development. It is hypothesized that the vascular plexus fails to regress in SWS.65,83 Fibronectin expression may play a role in this process.3,84
Pathologically, the normal vascular network of the brain is altered in SWS such that the cortical vessels are hypoplastic, the deep venous structures are usually dilated, and the leptomeningeal vessels are enlarged.59 Microscopic examination reveals calcium in the cortex with gliosis and hypoplastic vessels.65 Serial SPECT examinations demonstrate that blood supply to the affected cortex decreases progressively over the first year of life.85 Also, during seizures, blood flow is significantly decreased in the surrounding brain, creating a vascular steal phenomenon.68,86 Therefore, seizures may exacerbate ischemia.87
49.2.5 Diagnostic Studies
MR imaging is the radiologic test of choice for diagnosing leptomeningeal angiomatosis, which is visualized best on T1-weighted contrasted images (▶ Fig. 49.6). A choroid angioma with hypertrophy or dilation of the ipsilateral choroid plexus can be noted in older children. Newer MR imaging sequences, such as susceptibility-weighted imaging, are improving the diagnostic sensitivity of MR imaging by detecting the cortical calcification and dilated transmedullary and subependymal veins that may precede other MR imaging findings.88,89 Additionally, MR imaging perfusion and diffusion abnormalities have correlated with clinical findings such as hemiparesis, seizures, and cognitive performance.90–92 Computed tomography (CT) is also useful for demonstrating gyriform cortical and choroidal calcifications, which are often absent in infancy. Because of the vascular steal phenomenon, the affected part of the brain atrophies and the calvaria secondarily thickens.93,94
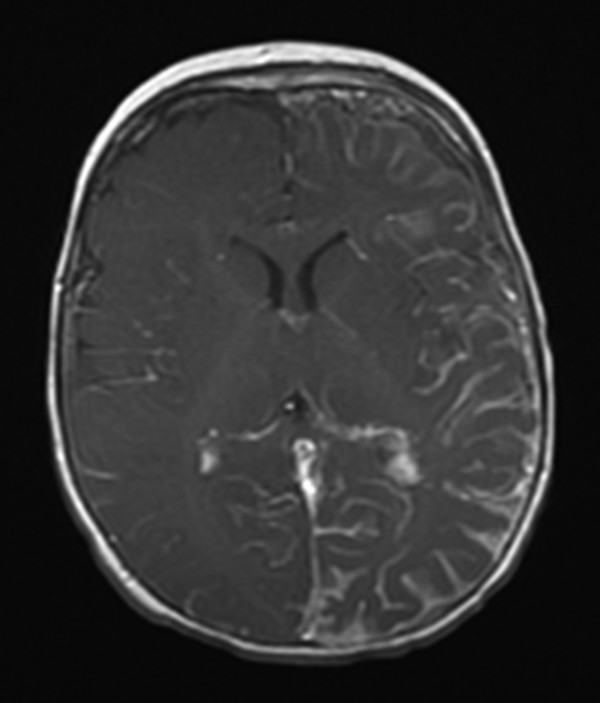
Fig. 49.6 Magnetic resonance (MR) imaging of the brain of an 11-month-old boy born with a left-sided port-wine stain in the distribution of cranial nerve V1. Initial MR imaging at 7 months was normal. On this axial T1-with-contrast sequence, left hemispheric leptomeningeal angiomatosis is demonstrated.
When patients present with strokelike episodes, they typically undergo CT (to rule out hemorrhage), EEG, and MR imaging. MR imaging may reveal diffusion abnormalities that are not in a large-vessel distribution. These are believed to be due to microvascular stasis.75,95
In an infant with a PWS, leptomeningeal angiomatosis can be difficult to diagnose because neither CT nor MR imaging is sensitive in this age group.88 Imaging may need to be repeated between the ages of 1 and 2 years to rule out intracranial involvement. Generally, it can be assumed that if a child older than 1 year with a PWS is developing normally, without seizures, and has normal findings on neurologic examination and contrasted brain MR imaging, then leptomeningeal involvement is unlikely.59,88
Metabolic neuroimaging modalities, such as fluorodeoxyglucose F 18 PET, can assist in a comparison of glucose utilization in the affected and unaffected hemispheres and potentially in making decisions about the timing and technique of epilepsy surgery.75,96,97
49.2.6 Treatment
A multidisciplinary approach is required for the care of SWS patients. They often require the care of a neurologist, ophthalmologist, dermatologist, and potentially a neurosurgeon. Because of the risk for glaucoma, even in infants, an ophthalmologist should be involved in the care of children with SWS.75 Neurologists can treat migraines with the same strategies that are used for patients without SWS.75,98
Neurologists can medically control seizures in 40% of cases.70 When medical control cannot be obtained, surgery is considered. Because the neurocognitive and neurologic deficits can be progressive, determining the timing of surgery can be difficult. Surgeons differ on the optimal timing of the surgery. To prevent the adverse effects of continued seizures, some surgeons advocate early surgery.70,74,99 Others think it is better to be more selective because of the potential for gaining seizure control with AEDs and the possible complications of surgery.100
Multiple surgical techniques have been employed, but hemispherectomy is the most effective in obtaining long-term control, and surgeons have reported good functional and quality-of-life outcomes.70,101 Hemispherectomy was first performed for this condition by Falconer and Rushworth.62 It is not clear whether there is an advantage of functional versus anatomical hemispherectomy in this patient population. Hemispherectomy is a reasonable choice for children with intractable epilepsy, hemiparesis, and visual field deficit. The more difficult decision pertains to the child with intractable epilepsy but without significant hemiparesis or visual field deficit.75 Special considerations must be weighed regarding the child’s vision. If a patient has normal visual fields, then the postoperative visual field deficit from a hemispherectomy with the possibility of visual loss from glaucoma must be considered.59 Although not as successful, more limited resections should be considered in patients without significant deficits. Surgery can be considered for patients with bilateral disease if monitoring localizes the seizures predominantly to one hemisphere.102
The strokelike episodes are difficult to treat because their etiology is not completely understood, and the potential benefit of treating is debatable.75 Because of the proposed etiology of microvascular stasis for this phenomenon, low-dose aspirin has been used with reports of decreased strokelike episodes and decreased seizures.103,104
49.2.7 Prognosis
The prognosis depends on the severity of disease. There are no reasonable long-term studies reporting the lifetime prognosis for SWS patients. Neurocognitive and functional outcomes are the result of multiple factors, including seizures, AEDs, the vascular steal phenomenon, and cortical atrophy. Generally, patients who have better seizure control, either with AEDs or through surgery, have better outcomes.59,91
49.3 von Hippel-Lindau Disease
49.3.1 Introduction
von Hippel-Lindau disease (VHL) is an autosomal-dominant genetic disorder that causes multiple neoplasms, including central nervous system (CNS) and retinal hemangioblastomas, pheochromocytomas, clear cell renal carcinoma, endolymphatic sac tumors, and pancreatic islet cell tumors. Renal and pancreatic cysts as well as epididymal and broad ligament cystadenomas can form.105,106
In 1904, Eugene von Hippel, an ophthalmologist, described familial retinal angiomas,107 later found to be histologically hemangioblastomas.108 In 1926, Arvid Lindau, a pathologist, noted the association of these retinal hemangioblastomas and cerebellar hemangioblastomas.109 There are now diagnostic criteria based on family history and clinical findings.107
Although VHL-associated neoplasms develop relatively early compared with their sporadic counterparts, VHL remains primarily an adult disease.110 However, children can present with VHL.111,112
49.3.2 Epidemiology
The incidence of VHL is 1 in 30,000 to 1 in 50,000 live births.113,114 It affects both sexes equally and can be found in all ethnic groups. There is a 90% penetrance by the age of 65.115 VHL accounts for a third of all CNS hemangioblastomas, more than half of retinal hemangioblastomas, and 1% of RCCs in the general population.116
49.3.3 Clinical Presentation
CNS hemangioblastoma is responsible for the presenting symptoms of 40% of patients at the time of VHL diagnosis.105 These vascular tumors ultimately manifest in 60 to 80% of patients with VHL.105,117 Overall, VHL causes 5 to 30% of all cranial hemangioblastomas and 80% of all spinal hemangioblastomas.118 The average age at a diagnosis of CNS hemangioblastoma is 29 years, although childhood cases are by no means rare.110 These tumors are most commonly located in the posterior fossa (75%), predominantly in the cerebellum but occasionally in the brainstem, and in the spinal cord (25%).119,120 The presenting symptoms are related to local mass effect, which can differ depending on whether the tumor is in the cerebellum, brainstem, or spinal cord. Because of this mass effect, posterior fossa hemangioblastomas often cause hydrocephalus. Additionally, the hemangioblastomas cause polycythemia in 5 to 20% of patients.118 Rarely, CNS hemangioblastomas are located in the supratentorial compartment.121
Retinal hemangioblastomas are the most common finding at presentation, and in half of cases, lesions are multiple or bilateral.122 By 50 years of age, the incidence of loss of vision is 35% in all VHL gene carriers and 55% in patients diagnosed with a retinal hemangioblastoma. Only 5% of cases are diagnosed before the age of 10 years.123
Pheochromocytomas are important for neurosurgeons to identify because of their potential effect on operative conditions and postoperative care.124 The incidence varies depending on the type of VHL (discussed below), but in most types the incidence ranges from 8 to 17%.108 Pheochromocytomas can cause palpitations, unstable blood pressure, sweating, and headaches. Unlike most other VHL neoplasms, they are more likely to be detected in children and young adults than in older patients.125
Endolymphatic sac tumors are present in 11% of patients and are usually asymptomatic at the time of diagnosis. When they do cause symptoms, hearing loss is usually the chief complaint, but they can also cause tinnitus and vertigo. With a mean age at diagnosis of 22 years, they can affect children.126
The kidneys are affected with both clear cell renal carcinoma and renal cysts, but neither usually causes the initial presenting symptoms of VHL.118,127 Although the incidence of RCC varies depending on the type of VHL, in the most common types, the lifetime risk is about 70%.110 VHL is the most common cause of hereditary kidney cancer.108 The average age at presentation is 40 years128 however, asymptomatic lesions are frequently diagnosed earlier, but rarely before the age of 16 years.129
Islet cell tumors and cysts affect the pancreas. Usually, the islet cell tumors are nonsecreting, and the cysts do not normally affect function.130
49.3.4 Pathology and Genetics
Inheritance is autosomal-dominant, with about 20% of cases sporadic.118 The VHL tumor suppressor gene was mapped to chromosome 3p25.131 Many gene mutations have been discovered, creating a complicated genotype–phenotype relationship.132 Certain genotypes are more likely to be associated with specific tumors, such as pheochromocytomas, which has led to the definition of different “types” of VHL.116,133 The mutations can be missense, nonsense, or deletions.134,135 Prenatal testing is available.136
Histologically, CNS and retinal hemangioblastomas are identical. These tumors are benign and highly vascular, consisting of large, lipid-laden stromal cells supported by a well-developed capillary network with many mast cells. The stromal cells show hyperchromasia and atypia, but almost no mitoses. The cyst fluid has a high concentration of erythropoietin.108,137
49.3.5 Diagnostic Studies
In 1964, clinical diagnostic criteria were first proposed by Melmon and Rosen after review of the literature.107 In a patient with a positive family history, the diagnosis is made if a single VHL-associated tumor is discovered. In the 20% of patients with sporadic VHL, either two or more CNS or retinal hemangioblastomas or a single hemangioblastoma and one other VHL-associated neoplasm must be present.107,134
On CT and MR imaging, CNS hemangioblastomas have characteristic findings, which include a cystic component and an enhancing mural nodule (▶ Fig. 49.7). Spinal cord hemangioblastomas have an enhancing intramedullary component, often with a cystic component and associated syrinx. CNS hemangioblastomas often have large feeding and draining vessels.138 The differential diagnosis can include juvenile pilocytic astrocytoma, ependymoma, and arteriovenous malformation.118,120,139
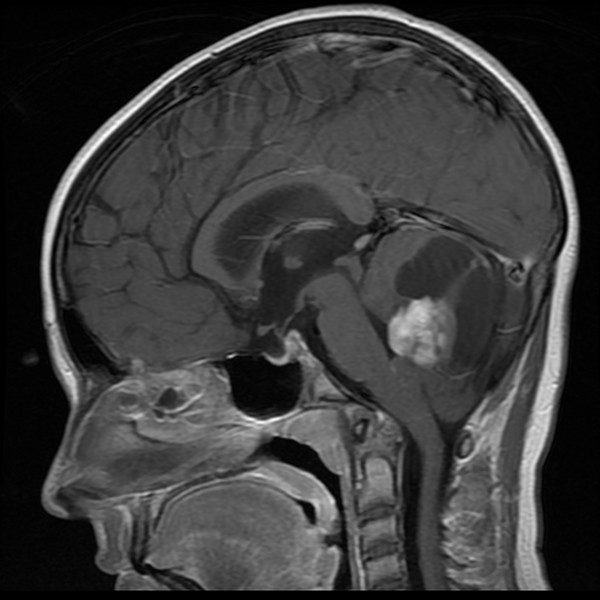
Fig. 49.7 Magnetic resonance imaging of the brain of a 10-year-old with von Hippel-Lindau disease. On this sagittal T1-with-contrast sequence, a posterior fossa hemangioblastoma is present, with the characteristic cystic component and enhancing mural nodule.
Endolymphatic sac tumors are best diagnosed on MR imaging of the brain with and without contrast to evaluate for an enhancing mass, and on thin-cut CT through the temporal bone to evaluate for bone erosion associated with these tumors. Sometimes, these tumors can be occult on imaging.140,141
CT and MR imaging of the abdomen can assist with the diagnosis of pheochromocytoma; however, laboratory tests are the gold standard. The diagnosis can be made with the measurement of either plasma fractionated metanephrines in high-risk patients or 24-hour urine fractionated or total metanephrines and catecholamines in lower-risk patients.142
Screening recommendations have been proposed by Maher et al.116 Patients with VHL should undergo screening MR imaging of the brain and possibly spine every 1 to 3 years beginning in adolescence. An ophthalmologist should screen for retinal hemangioblastomas annually beginning in infancy or childhood. Annual renal and pancreatic screening MR imaging or ultrasound should begin at the age of 16 years. For pheochromocytoma, blood pressure monitoring and 24-hour urine catecholamine metabolite studies should be performed annually beginning in childhood. In families at higher risk for pheochromocytoma, more intense surveillance, including annual plasma normetanephrine level measurement and adrenal imaging beginning at the age of 8 years, should be considered.
49.3.6 Treatment
Natural history studies of CNS hemangioblastomas describe saltatory growth patterns with intermittent, unpredictable periods of quiescence and growth.112,117,143 Not all tumors cause symptoms. In VHL, it is recommended that tumors be treated only when they are causing symptoms.112,141,143,144 Symptomatic tumors almost universally are associated with either cysts or peritumoral edema.144 At this time, there is no way to predict which tumors will progress to cause symptoms and require surgical treatment.
Angiography can be useful for defining the vascular anatomy. Preoperative embolization has been used; however, it is often not possible because much of the blood supply may be provided by multiple small vessels that cannot be embolized.141 Also, there have been reports of significant morbidity and mortality associated with embolization of these tumors.145,146
Surgical resection of symptomatic CNS hemangioblastomas is the recommended therapy. Because of the vascularity, careful microsurgical technique must be employed. Extracapsular dissection with careful coagulation and transection of tumor vessels is recommended.144,147,148 Disruption of the tumor capsule can cause troublesome bleeding. When there is an associated cyst, the cyst capsule is maintained as long as possible to assist with dissection. The cyst wall does not require resection because it is only compressed gliotic tissue.149
A similar approach should be taken with spinal cord hemangioblastomas. In symptomatic lesions, surgical resection should be undertaken. Poor outcomes have been associated with a ventral location and not presenting to the pial surface.150
Other treatment strategies include stereotactic radiation, with reports of short-term tumor control over 90%.151 However, at 15 years of follow-up, tumor control decreases to 51%.152
In patients requiring surgical treatment of a CNS hemangioblastoma who also harbor a pheochromocytoma, alpha- and beta-adrenergic blockade must be employed preoperatively to safely manage the blood pressure perioperatively.153
The treatment of other tumors associated with VHL is beyond the scope of this chapter.
49.3.7 Prognosis
The early diagnosis of retinal hemangioblastomas and RCCs has improved patient outcomes.110 The most common causes of death are RCC and complications of CNS hemangioblastomas. The median life expectancy is 49 years.118,154 Because of the availability of genetic testing and the potential for early diagnosis in familial cases, life expectancy may potentially be extended with early diagnosis and treatment.136
49.4 Hereditary Hemorrhagic Telangiectasia
49.4.1 Introduction
Hereditary hemorrhagic telangiectasia (HHT), also known as Osler-Weber-Rendu syndrome, is one of the most prevalent autosomal-dominant conditions; there are only rare sporadic cases. The incidence is estimated to be 1 in 5,000 live births.155 Genetic testing is available and is positive in 85% of cases.156 The genetic mutations causing HHT have been linked to chromosomes 9q,157 12q,158 5q,159 18q, and 7p.160
The syndrome is characterized by visible cutaneous telangiectases, mucosal telangiectases causing recurrent epistaxis and gastrointestinal bleeding, and the potential for multiple-organ arteriovenous malformations (AVMs).161 Telangiectases are usually located on the lips, tongue, face, fingers, and mucosa. They are pink to red pinhead-size lesions, or sometimes elevated, purple lesions. They can be differentiated from petechiae because they blanch with pressure and then immediately refill.162 The cutaneous stigmata of HHT are not present at birth but evolve and progress with age.163 AVMs can form in the brain, spine, lungs, and liver. These patients also have higher incidences of anemia and thomboemboli due to elevated factor VIII levels, which occur in nearly 7% of patients.164 The average age at the onset of epistaxis is 12 years.163 Penetrance is age-related, approaching 100% by age 40.155
The diagnosis is made with the Curaçao criteria, which include the presence of epistaxis, telangiectases, visceral lesions, and family history. Patients are given the diagnosis of unlikely, possible, or definite HHT depending on the number of clinical criteria that are met.165 Genetic testing can be used to define the specific mutation in a family and then test other members of the family who may not meet the diagnostic criteria. Prenatal genetic testing can be performed for affected families. However, the benefit of such diagnosis has not been defined.166
Although there is no definitive life expectancy study, a number of reports indicate that life expectancy is likely lower for all patients with HHT because of the risks for death early in life from cerebral AVMs167,168 and for pregnancy-related maternal deaths; there is a 1% risk for maternal or fetal death with pregnancy.169 Five different types of HHT have been identified, and each is associated with different risk for certain AVMs.170
49.4.2 Neurosurgical Implications
Up to 23% of patients with HHT also have some type of cerebral vascular malformation. Most commonly, these lesions are AVMs, arteriovenous fistulas, cavernous malformations, and telangiectases.171 Current HHT guidelines recommend that children and adults with possible or definite HHT (by the Curaçao criteria) be screened with MR imaging. Patients with an AVM diagnosed on MR imaging should be referred for more specific testing and treatment.166 If a person’s childhood screening MR imaging study is negative for a vascular malformation, there is insufficient evidence to recommend regular follow-up screening with MR imaging; however, consideration should be given to another single screening MR imaging study at the beginning of adulthood.166 AVMs should be treated by neurosurgeons using the same clinical approach that they would use for sporadic AVMs, including the possibilities of microsurgery, stereotactic radiation, and embolization.172–174
Pulmonary AVMs occur in 15 to 50% of patients, and in addition to respiratory complications, there are multiple potential neurologic sequelae.175–177 Because of paradoxical embolism with right-to-left cardiovascular shunt, patients are at risk for transient ischemic attacks, ischemic strokes, and cerebral abscesses.177 In fact, neurologic symptoms in HHT are more likely to be due to an ischemic stroke than to a hemorrhage from a cerebral AVM.167,177
Fewer than 1% of patients develop spinal AVMs, which may present with paraparesis, paraplegia, myelopathy, or pain.18,19 No specific screening recommendations for spinal AVMs exist, but if an AVM is suspected, then MR imaging can be performed.166 Treatment depends on the type of spinal AVM but can include embolization or microsurgery.178
49.5 Basal Cell Nevus Syndrome
49.5.1 Introduction
Basal cell nevus syndrome (BCNS) is also known as Gorlin syndrome, Gorlin-Goltz syndrome, nevoid basal cell carcinoma, and basal cell carcinoma nevus syndrome. It has autosomal-dominant inheritance and is associated with the formation of basal cell carcinomas, odontogenic keratocysts, and skeletal anomalies. It was first defined as a syndrome in 1960 by Drs. Gorlin and Goltz.179 The association between BCNS and medulloblastoma was first described by Herzberg and Wiskemann in 1963.180 BCNS occurs in about 1 in 57,000 live births.181 The genetic mutations have been identified in the sonic hedgehog pathway.182,183 According to a consensus statement,184 a diagnosis of BCNS can be reasonably considered if any of the following criteria are met: one major criterion and molecular confirmation, two major criteria, or one major and two minor criteria (see box “▶ Diagnostic Criteria for Basal Cell Nevus Syndrome”).
Diagnostic Criteria for Basal Cell Nevus Syndrome
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


