The Principles of Psychopharmacology
Classification of psychotropic drugs
The classification of psychotropic drugs followed in this book is similar to that used by the British National Formulary. It is given together with the relevant chapter numbers from this book.
Antipsychotic drugs:
non-depot antipsychotics (typical, e.g. chlorpromazine, and atypical, e.g. aripiprazole) (Chapter 3)
depot antipsychotics, e.g. flupentixol decanoate (Chapter 4).
Antimanic drugs—e.g. lithium salts, carbamazepine, and valproic acid (Chapter 6).
Antidepressants:
tricyclic and related antidepressants, e.g. amitriptyline (Chapter 7)
monoamine-oxidase inhibitors, e.g. moclobemide (Chapter 8)
selective serotonin re-uptake inhibitors, e.g. escitalopram (Chapter 9)
other antidepressants, e.g. agomelatine (Chapter 10).
Hypnotics—e.g. zaleplon (Chapter 12).
Anxiolytics—e.g. lorazepam (Chapter 12).
Barbiturates (Chapter 12).
Central nervous system stimulants—e.g. atomoxetine (Chapter 12).
Antimuscarinic drugs used in parkinsonism—e.g. procyclidine (Chapter 12).
Drugs used in substance dependence—e.g. acamprosate (Chapter 12).
Drugs for dementia—e.g. memantine (Chapter 12).
The principles of absorption, distribution, metabolism, and elimination of drugs
When a psychotropic drug is administered to a patient, the pharmacokinetic processes relating to the drug refer to the effects of his body on the actions of this medication and include the processes of its absorption, distribution, metabolism and elimination. For most drugs, systemic circulation drug concentration is related to drug concentration at its sites of action, as shown in Fig. 2.1. By knowing the pharmacokinetics of a drug, together with the drug dose taken, it is possible to calculate drug concentration as a function of time at the sites of drug action.
Absorption
The processes by means of which a drug leaves its site of administration and enters the systemic circulation are referred to as drug absorption. The main types of absorption can be classified as follows:
Enteral administration—drug absorption into the systemic circulation from the gastrointestinal tract:
oral
buccal or sublingual
rectal.
Parenteral administration—drug absorption into the systemic circulation from sites other than the gastrointestinal tract:
intramuscular
intravenous
subcutaneous
inhalational
topical
intranasal
intra-arterial
intrathecal.
Factors which affect the rate of drug absorption include:
The form of the drug:
enteric coating, for example, slows down drug disintegration in the stomach.
The solubility of the drug, which depends on:
the pKa of the drug, which is the pH at which precisely half of the drug is in its ionized form
the size of particles in the formulation
the pH.
The rate of blood flow at the site of administration:
the higher the blood flow, the greater the rate of drug absorption.
Distribution
The distribution of a drug follows its absorption, or direct administration, into the systemic blood circulation and refers to the way in which it is
distributed between the lipid, protein, and water compartments. Factors which influence drug distribution include the following:
distributed between the lipid, protein, and water compartments. Factors which influence drug distribution include the following:
 Fig. 2.1 The relationship between drug pharmacokinetics and the concentration of the drug in the systemic circulation and at its sites of action. |
Haemodynamic factors:
cardiac output.
regional blood flow—those organs with the highest blood perfusion rates, such as (under normal circumstances) the brain, liver, and kidneys, receive the highest distribution to begin with, while a second distribution phase then gradually sees a redistribution to tissues such as resting skeletal muscle, adipose tissue, most viscera and the skin.
Plasma-protein binding. This acts as a drug reservoir since pharmacological activity, if it exists, resides with the free (unbound) drug fraction. As shown in Fig. 2.1, the free fraction and the protein-bound fraction are in equilibrium with each other. Acidic drugs mainly bind to albumin. Many psychotropic drugs are basic, and they may bind to, for example, α1-acid glycoprotein and lipoprotein. Plasma-protein binding is usually reversible (not covalent). Factors that can alter the degree of plasma-protein binding include:
plasma drug concentration—the usual relationship between plasma-protein binding and plasma drug concentration is that shown in Fig. 2.2
hypoalbuminaemia—for instance as a result of hepatic or renal disease, cardiac failure, malnutrition, carcinoma, surgery, or burns
acute phase response—disorders causing this, including myocardial infarction, carcinoma, arthritis, and Crohn’s disease, can raise the
plasma level of the acute-phase reactant α1-acid glycoprotein (and therefore increased plasma-protein binding of basic drugs)
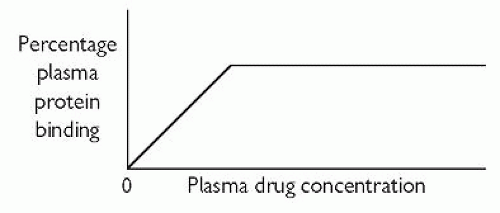
Fig. 2.2 The usual relationship between plasma-protein binding and plasma drug concentration. Reproduced with permission from Puri BK and Tyer PJ (1998). Sciences Basic to Psychiatry, 2nd edn. Churchill Livingstone, Edinburgh.
hypo-α1-acid glycoproteinaemia—which may occur in the nephrotic syndrome, hepatic disease and malnutrition
hyperlipoproteinaemia—types IIa, IIb, and IV
drug interaction
changes in the concentration of physiological substances—such as urea, bilirubin and free fatty acids.
Permeability factors. In general, the higher the lipid-solubility of a drug, the greater its rate of entry into cells.
The blood-brain barrier. This acts as a functional barrier between the cerebral circulation and the brain and is formed from:
the tight junctions between adjacent endothelial cells
the gliovascular membrane (formed from astrocytic perivascular feet on capillaries)
the capillary basement membrane
membrane transporters acting as efflux carriers—particularly P-glycoprotein.
In general, the higher the lipid-solubility of a drug, the greater the probability that it will cross the blood-brain barrier. The blood-brain barrier may have altered properties in the presence of infection. (For instance, while normally penicillins, being highly water-soluble, do not readily cross the blood-brain barrier, during acute bacterial meningitis this situation changes markedly.) Active transport mechanisms are used to gain entry into the central nervous system by certain substances, such as levodopa. Note that small molecules (e.g. lithium ions) tend to be able to diffuse into the brain (and cerebrospinal fluid) from the systemic circulation.
The blood-cerebrospinal fluid barrier. This is similar to the blood-brain barrier and exists at the choroid plexus. The tight junctions in the blood-cerebrospinal fluid barrier are between adjacent epithelial cells (as opposed to adjacent endothelial cells in the case of the blood-brain barrier).
The placenta. Drug transfer across the placenta from the maternal circulation to the fetal circulation may take place via:
passive diffusion
active transport
pinocytosis.
In general, it should be assumed that there is fetal exposure to any drugs or food supplements taken by the mother during pregnancy. Placental drug transfer is of importance throughout pregnancy owing the distinct possibility of adverse effects on the growing fetus or baby:
first trimester—teratogenesis may result (the highest risk period is between the 3rd and 11th weeks). If possible, all drugs and food supplements should be avoided during the first trimester. If a drug has to be given during this time, the smallest effective dose should be used and preference should be given to using a drug that has a history of having been used apparently safely in pregnancy rather than a newer drug or a drug that has no well-established history of safe use during pregnancy.
second and third trimesters—drugs may affect fetal growth and development. Toxic side-effects may occur in fetal tissues.
parturition—drugs given during labour may affect the neonate.
Metabolism
The metabolism or biotransformation of a drug generally renders it less lipid-soluble and more water-soluble. The products of such metabolism are more readily eliminated from the body. These products are not necessarily pharmacologically inactive. For instance, the serotonin re-uptake inhibitor fluoxetine is metabolized to the active drug norfluoxetine, while the benzodiazepine diazepam is metabolized to the active drug nordiazepam. Similarly, paliperidone (9-hydroxyrisperidone) is an active metabolite of risperidone.
Some highly water-soluble drugs, such as lithium salts, are excreted unchanged by the body.
The major site of drug metabolism is the liver. Other sites of significant biotransformation (which probably occurs in all tissues) are the:
gastrointestinal tract
kidneys
lungs
suprarenal (adrenal) cortex
placenta
skin
lymphocytes.
Two phases are broadly defined for hepatic biotransformation, as shown in Fig. 2.3.
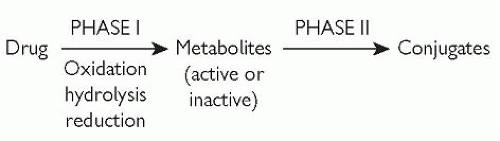 Fig. 2.3 Phase I and phase II metabolism (biotransformation). Reproduced with permission from Puri BK and Tyer PJ (1998). Sciences Basic to Psychiatry, 2nd edn. Churchill Livingstone, Edinburgh. |
Phase I metabolism causes molecular structure changes by means of one or more of the following non-synthetic reactions:
Oxidation—by microsomal mixed-function oxidases using the cytochrome P450 monooxygenase system, particularly CYP3A4, CYP3A5, CYP2C, and CYP2D6. Oxidative reactions and examples of drugs metabolized by them include:
aliphatic hydroxylation—e.g. meprobamate, pentobarbital
aromatic hydroxylation—e.g. amphetamine, phenytoin, propranolol
N-dealkylation (N-demethylation)—e.g. diazepam, amitriptyline, imipramine
O-dealkylation (O-demethylation)—e.g. codeine
S-dealkylation (S-demethylation)—e.g. 6-methyl thiopurine
deamination—e.g. amphetamine, diazepam
S-oxidation—e.g. chlorpromazine
Epoxidation—e.g. carbamazepine, phenytoin.
Hydrolysis—in the smooth endoplasmic reticulum; it is not a common phase I reaction (e.g. pethidine).
Reduction—in the smooth endoplasmic reticulum; it is not a common phase I reaction.
A drug or drug metabolite (from a phase I reaction) is conjugated to a polar (water-soluble) group by phase II metabolism. The result is a water-soluble conjugate which can undergo renal excretion relatively easily if it has a relative molecular mass (molecular weight) of less than approximately 300. If the relative molecular mass is much higher than 300, excretion can occur via the bile.
Conjugation reactions and examples of corresponding drug substrates include conjugation with:
glucuronic acid—e.g. lorazepam, oxazepam, phenytoin
sulphate—e.g. estrone
acetate—e.g. hydralazine
glutathione—e.g. metabolites such as epoxides
glycine—e.g. salicylates.
Phase I and phase II metabolic enzyme activity may be increased by certain drugs. In turn, this can result in increased metabolism of these drugs themselves and/or other drugs. Examples of drugs that cause enzyme induction include:
alcohol (ethanol)—induces increased metabolism of alcohol, barbiturates, bilirubin, meprobamate and phenytoin
barbiturates—induce increased metabolism of barbiturates, chlorpromazine, the oral contraceptive pill, phenytoin, tricyclic antidepressants, etc.
carbamazepine—induces increased metabolism of carbamazepine
phenothiazines—induce increased metabolism of phenothiazines.
Note that enzyme induction does not necessarily reduce the risk of adverse side-effects. For example, microsomal enzyme induction by chronic alcohol (ethanol) intake can lead to increased levels of potentially hepatotoxic paracetamol phase I metabolites.
Drug metabolism that an orally administered drug undergoes by the liver or gut wall before entry into the systemic circulation is the first-pass
effect (also known as first-pass metabolism, first-pass elimination, presystemic metabolism, and presystemic elimination). The result can be a substantial reduction in drug bioavailability, so that a higher oral dose needs to be given. Furthermore, there may be a high degree of variation in the level of the first-pass effect between different individuals.
effect (also known as first-pass metabolism, first-pass elimination, presystemic metabolism, and presystemic elimination). The result can be a substantial reduction in drug bioavailability, so that a higher oral dose needs to be given. Furthermore, there may be a high degree of variation in the level of the first-pass effect between different individuals.
Examples of drugs which undergo a high degree of first-pass metabolism include:
imipramine—only 30-80% of the oral dose enters the systemic circulation intact
fluphenazine—only around 10% of the oral dose enters the systemic circulation intact.
The level of the first-pass effect may be reduced in a number of ways:
increased hepatic blood flow—following the ingestion of food or after taking a drug such as hydralazine
hepatic impairment—e.g. as a result of hepatic cirrhosis.
Elimination
The major organ of drug excretion is the kidney. Drug excretion can also occur though:
bile and faeces
lungs
saliva
sweat
sebum
tears
milk.
The key processes involved in the renal excretion of drugs are:
Glomerular filtration. This amount of drug that enters the glomerular filtrate depends on factors such as the:
glomerular filtration rate
plasma water drug concentration
relative molecular mass of the drug.
Active tubular secretion from the plasma to the glomerular filtrate of charged molecules in the proximal renal tubule. There are 2 main systems, for:
organic acids—e.g. glucuronide drug metabolites
organic bases—e.g. amphetamine.
Passive tubular reabsorption from the glomerular filtrate to the blood, since the former becomes increasingly concentrated as it passes along the nephron. The extent of this reabsorption varies with drug/metabolite lipid solubility, which in turn varies with the:
pKa of the drug or metabolite
pH of the tubular urine—e.g. reduced reabsorption occurs in alkaline tubular urine of acidic drugs, hence the use of forced alkaline diuresis in cases of aspirin (salicylate) and phenobarbitone overdose.
Conditions causing reduced renal clearance (e.g. renal disease) can reduce the excretion of drugs such as lithium salts, thereby resulting in lithium toxicity.
Cycling processes are processes in which, following excretion, some drugs may be reabsorbed into the systemic circulation, thereby being recycled. Examples of cycling processes include:
the enterohepatic cycle—reabsorption of a drug (or drug metabolite) from the intestinal tract following hepatic excretion in bile
gastric excretion—reabsorption of a drug (or drug metabolite) from the intestinal tract following gastric excretion
salivary excretion—reabsorption of a drug (or drug metabolite) from the intestinal tract following salivary excretion.
Pharmacokinetic terms and formulae
Volume of distribution
This theoretical entity is the quotient of the drug dose (or drug mass), D, in the body to the plasma or blood concentration, C, of the drug at that time. The volume of distribution, Vd, is given by:
Therefore the volume of distribution is the theoretical blood or plasma volume needed to account for the drug dose taken, to give the measured drug concentration. The volume of distribution does not usually correspond to either an anatomical or a physiological volume.
Half-life
Following intravenous injection, there is a rapid fall in the plasma drug concentration. This is caused by redistribution of the drug from the blood circulation into other tissues. The time taken for this redistribution process to halve the initial peak drug concentration is the distribution half-life, as shown in Fig. 2.4. As indicated in this diagram, the redistribution process is usually a rapid process, with a correspondingly short half-life.
Following redistribution, the slower process of drug elimination becomes increasingly evident. The time taken by this elimination process to halve the plasma drug concentration is the elimination half-life, also shown in Fig. 2.4.
Suppose a drug were being administered by continuous intravenous infusion. If the plasma concentration of the drug reached a constant value of C0, and if the elimination half-life were constant at t½, then, ignoring any redistribution effects, upon suddenly stopping the infusion (at time zero) the plasma drug concentrations at integer (whole number) multiples of the half-life would be as shown in Table 2.1.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


