Fig. 12.1
Minerals and trace elements: relationship with cognition, brain function, Parkinson and Alzheimer disease and other diseases such as Friedreich ataxia
Consideration should be taken into account regarding the handling of trace elements. Because micronutrient deficiencies often coexist and synergistic effects of micronutrients on physical functions may indirectly affect cognition, supplementing children and elderly people with multiple micronutrients could have advantages over single micronutrient supplementation. In contrast, micronutrients might also have antagonistic effects, affecting their bioavailability and their functioning in physiologic processes which could lead to impaired cognitive functioning. Because iron and zinc and copper and manganese compete for intestinal uptake, a high dose of one of these minerals may limit the absorption of the others [7].
Iron
Observational studies have found relationships between iron-deficiency anemia in children and poor cognitive development, poor school achievement, and behavior problems. However, it is difficult to separate the effects of ID anemia (IDA) from other types of deprivation in such studies, and confounding factors may contribute to the association between ID and cognitive deficits [8].
Several possible mechanisms link IDA to altered cognition. Anemic children tend to move around and explore their environment less than children without anemia, which can lead to developmental delays [9].
Conduction of auditory and optic nerve impulses to the brain has been found to be slower in children with IDA. This effect could be associated with changes in nerve myelination, which have been observed in iron-deficient animals [10]. Neurotransmitter synthesis may also be sensitive to ID [11]. Impaired intellectual development in children can be prevented through the treatment or prevention of ID.
On the other hand, iron overload can be related to neurodegenerative disease. Several genetic disorders can lead to pathological accumulation of iron in the body; the body’s tight control of intestinal iron absorption protects it from the adverse effects of iron overload [12]. Iron is required for normal brain and nerve function through its involvement in cellular metabolism and in the synthesis of neurotransmitters and myelin.
However, accumulation of iron excess can result in increased oxidative stress, and the brain is particularly susceptible to oxidative damage. Iron accumulation and oxidative injury are currently under consideration as potential contributors to a number of neurodegenerative diseases such as AD and PD [13, 14].
The abnormal accumulation of iron in the brain does not appear to be a result of increased dietary iron, but rather a disruption in the complex process of cellular iron regulation. Although the mechanisms for this disruption in iron regulation are not yet known, it is currently an active area of biomedical research.
Iron in the Brain
The iron needs of the brain vary with the stage of the life cycle and the cell types in the CNS. Iron is the key component of the many enzymes that involve essential oxidation/reduction reactions, synthesis of neurotransmitters, catabolism of neurotransmitters, and synthetic processes such as the production of myelin.
The highest levels of iron in the brain are found in the basal ganglia, but iron is also found throughout the brain, including the white matter [15]. Both biochemical and histochemical studies reveal white matter throughout the brain as a major site of iron concentration. Iron uptake into the brain is at a maximum during the period of rapid brain growth, coinciding with the peak of myelinogenesis.
Lack of iron in the diet for the first 2 years of life has an important effect on development because that is the time when the majority of brain growth occurs. Although the most rapid brain growth is seen in the months leading up to birth, at birth the brain has only reached 27 % of its adult size and it continues to grow for the next 2 years [16]. It has been shown that iron levels in the brain at birth are 10 % of eventual adult levels, with the remainder accumulating through childhood and young adulthood [15].
Research in humans was directed to impaired iron transport across the placenta in several prenatal conditions (e.g., diabetes mellitus, prenatal alcohol exposure, intrauterine growth retardation, maternal stress). There is direct evidence of decreased brain iron or ID in the offspring son and daughter [17].
Infants born of mothers with nutritional ID during pregnancy are rarely anemic, but they may have lower iron stores and ID sooner in the postnatal period. There is now solid evidence that brain ID can occur even with a normal hemoglobin level.
Brain iron accumulates from birth to early adulthood [15]. Perhaps for this reason, brain iron levels are affected more seriously by ID in the very young than in the adult animal. A brief period of severe ID in the young rat, but not in the adult, resulted in a deficit of brain iron which was not corrected by iron therapy although all signs of systemic ID were reversed [18].
This resistance contrasts with the rapid normalization of hepatic iron and hemoglobin (Hb) concentrations following iron repletion. The failure to reverse iron depletion in the brain with iron treatment seems to be the result of a slow rate of replacement of brain iron compounds [19].
The development of iron-deficient animal models has been an invaluable tool for looking at the consequences of ID. Most animal studies have involved rats because the distribution of iron in their brain is comparable with that of the human brain. Animal studies have shown that ID is associated with hypomyelination of neurons in the developing brain [20].
It must be recognized, however, that there are certain limitations in using animal models. Rats are less mature at birth, and humans and rats have different rates of neuronal development, both of which are particularly important when assessing the effect of early iron deprivation on mental function [21].
There has been considerable research on the possible mechanisms through which IDA affects cognitive function. Most emphasis has been placed on a direct neurochemical effect. ID causes low levels of brain iron, which leads to a reduction in neurotransmitter levels, impaired transmitter function, hypomyelination, and delayed neuromaturation. Another possibility is that the systemic effects of anemia lead to low oxygen delivery to the brain, directly affecting cognition [22].
When ID occurs early in the development of the rat, there are lasting deficits in brain iron, electrophysiological changes, a decrease in the number of dopamine D2 receptors, and alterations in neurotransmitter function, hypomyelination, and persisting behavioral changes that suggest an altered threshold of arousal [23].
The second hypothesis for the mechanism is that ID also has an indirect effect on behavior. IDA infants and children have been shown to be less attentive and less responsive. Lower developmental scores may reflect poorer mother/child interaction because of the child’s reduced responsiveness, and this could result in less effective stimulation of the environment. The lower scores can also reflect poorer interaction with the developmental assessor [24].
To specifically understand the role of iron in neural circuits that underlie learning and memory development and function, in more recent models, iron uptake genes have been genetically manipulated in a tissue- and time-specific manner to generate a non-anemic model of hippocampal ID. The models are capable of isolating the role of iron independent of the potential widespread confounding effects of brain and body ID that accompany maternal dietary restriction (e.g., hypoxia, uptake of other divalent metals [25].
Optimal neurodevelopment is shaped by a variety of factors including growth factors, synaptic activity, and environment. Structures are most sensitive to these factors during rapid development [26].
As noted above, humans are most vulnerable to early ID from late gestation through 2–3 years of age, during the most rapid period of hippocampal structural maturation. Functionally, hippocampus-dependent memory appears and matures between 3 and 18 months of age [27].
This increased metabolic activity is coincident with extensive dendrite arborization, spine formation, and synaptogenesis [28] as well as the maturation of electrophysiological plasticity [29]. In conjunction, the timing and energy demands of hippocampal development with long-term deficits support the vulnerability of the structure to the metabolic consequences of early-life ID [30].
The effects of early-life ID on hippocampus-based learning and memory have been largely ascribed to primary abnormalities in iron-containing proteins, although many effects can be attributed to iron-containing proteins (e.g., reduced neuronal energy capacity).
Iron is necessary for energy production and cellular metabolism because it is essential for many mitochondrial enzymes integral for oxidative phosphorylation and adenosine triphosphate (ATP) production, including cytochromes, Nicotinamide adenine dinucleotide phosphate, and flavoproteins [31]. Adequate energy availability is necessary to support neuronal development and synaptic activity.
At birth, the brain comprises 50 % of resting metabolic energy [32, 33]. Approximately one half of the energy consumption is used to maintain Na+, K+, and Ca2+ gradients necessary for generating membrane potentials required for synaptic transmission. In addition, the generation and maintenance of the complex neuronal structure requires large amounts of energy.
Another important cellular process dependent on iron availability is nucleic acid metabolism. Iron-containing enzymes such as ribonucleotide reductase, deoxyribonucleic acid (DNA) helicase elongation protein 3, and BACH1 are integral for deoxynucleotide triphosphates (dNTP) synthesis, DNA transcription, elongation, and repair, and histone modification [34, 35].
The exact mechanism by which ID induces these acute and persistent gene expression changes is not clear because the experiments utilized maternal dietary restriction models of early IDA and the alterations may be due in part to the contribution of hypoxia. However, the evidence suggests that early life ID affects the regulation of gene expression throughout life in the hippocampus.
Another set of important signaling pathways that are likely affected by ID are found in mitochondria. The cellular functions of mitochondria reach beyond ATP synthesis and include maturation of Fe–S proteins that are crucial for cell function [36, 37]. As part of their function, mitochondria are an important factor in the regulation of intracellular Ca2+ levels. This function is crucial for many aspects of neuronal function, including secondary signaling cascades, neurotransmitter release, and apoptosis [38].
Iron Deficiency and Neural Functioning in Humans
Impaired Intellectual Development in Children
Infancy is considered the age range of highest vulnerability for the CNS because it corresponds with the brain growth spurt and the unfolding fundamental mental and motor processes. Altered behavior and development are among the greatest concerns regarding ID in infancy, especially because the nutrient deficiency is most prevalent in the period between 6 and 24 months of age. Because this age range coincides with a period of maximal brain growth and the unfolding of many neuron developmental processes, several investigators have focused on the question of CNS effects on ID [39].
In observational studies, anemia and ID are associated with cognitive deficits, suggesting that iron supplementation may improve cognitive function and the studies show cross-sectional associations between IDA and poor cognitive function, motor development, behaviour, or school achievement levels [9]. However, it is difficult to separate the effects of IDA from other types of deprivation in such studies, and confounding factors may contribute to the association between ID and cognitive deficits [40].
In observational studies, anemia and ID are associated with cognitive deficits, suggesting that iron supplementation may improve cognitive function. The effect of iron supplementation on a range of health outcomes in infants and young children has been well documented. It is estimated that 47 % of preschool children worldwide have anemia, the highest prevalence of any population group [41].
Longitudinal studies show that ID in infancy is related to poorer cognition in childhood [9]. One systematic review that included seven randomized controlled trials on the effects of supplementary iron in young children with anemia or ID found no evidence of an effect of iron supplementation on psychomotor development [42], while another included 17 randomized controlled trials in children of any age and with any initial iron status, found that iron supplementation was not associated with improved mental development scores in children younger than 5 years [43], or with improved physical growth [44].
A more recent systematic review addressed a range of health risks and benefits of iron supplementation in infants and children 5 years old or younger [45], finding that supplementation led to improvements in cognition and motor development in children with anemia and ID, but was associated with increased risk of death in areas with endemic malaria.
As previously explained deficiency of enzymes involved in the development of parts of the brain is important for cognitive functions such as memory (e.g. the hippocampus). Deficiency and supplementation may have different effects on infants and young children than in other population groups.
Older Children, Adolescents, and Adults
Older children and adolescents are less at risk of anemia than preschool children, but global statistics indicate that approximately 25 % of older children have anemia, as do 30 % of non-pregnant women and 42 % of pregnant women, and 17 % of elderly people (rising to 40–50 % of those admitted to hospital or living in nursing homes), demonstrating that it is a very large and important health problem [41, 46].
A meta-analysis has been published to assess whether iron supplementation improved cognitive domains: concentration, intelligence, memory, psychomotor skills, and scholastic achievement in adults. Evidence was found that iron supplementation improved attention and concentration in adolescents and women, regardless of baseline level of iron status. Iron supplementation also improved performance in intelligence quotient (IQ) tests in adults and children who were anemic at baseline, but had no effect in other groups or on other cognitive domains [47].
The prevalence of depleted iron stores is substantially greater in pre- or perimenopausal women than in postmenopausal women or in men. Poor iron status affects premenopausal women more often than men because of the combination of low dietary iron intake, menstruation, and gestational requirements. Physical performance is affected by poor iron status, including decreases in work productivity, voluntary activity, and athletic performance. Cognitive, affective, behavioral, and neurophysiologic decreases have been associated with poor iron status in premenopausal women [48].
It is important to consider that ID can often be present without anemia [41]. A requisite for successful treatment is the correct diagnosis of depletion and to assess the causes of ID [49].
Iron supplementation may be less effective when there are a number of nutritional problems at baseline (all of which may be contributing to cognitive limitations) than when patients are nutritionally replete except for variations in iron status. Iron and zinc deficiencies often occur together, and zinc deficiency can be exacerbated with a high dose of iron supplements [50]. Zinc may also play a role in cognitive function, therefore iron supplementation could exacerbate cognitive deficits [51].
Although it is not surprising that the brain functions poorly when iron is deficient, the long-term deficits, despite iron repletion, remain mechanistically enigmatic and a fruitful area of research. Furthermore, this research may contribute to defining the time point at which iron repletion can no longer reverse the behavioral phenotype. It would be critical in determining the optimal timing of iron treatment regimens.
Because iron is not only a critical nutrient for brain development but also a potentially toxic element, further research is also necessary to determine optimal iron doses. Arguably, an iron-deficient developing brain that has responded to ID by prematurely expressing large amounts of iron transporters [52] can be at risk for iron overload and generation of reactive oxygen species if large amounts of medicinal iron are suddenly delivered to this “activated system”.
Iron Overload and the Central Nervous System
Several proteins implicated in brain iron homeostasis are involved in disorders associated with abnormal iron metabolism. A basic understanding of mechanisms of iron homeostasis has a clinical relevance, as either accumulation or depletion of intracellular iron may impair normal function and promote cell death.
Iron accumulates in selective brain regions during aging, in acquired neurodegenerative disorders such as AD and PD, and in genetic disorders such as neurodegeneration with brain iron accumulation (NBIA). Dysregulation of iron homeostasis is also a critical feature of FA [53].
Iron is known to catalyze the formation of reactive oxygen species (ROS), such as hydroxyl radical, and initiation or enhancement of lipid peroxidation by reacting with hydrogen peroxide (H2O2) via the Fenton reaction [54]. ROS are highly reactive oxygen-containing radicals that can easily react with other molecules such as protein, DNA, lipids, and antioxidants [55, 56].
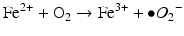
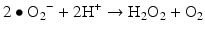
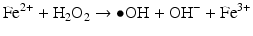 Iron can also react with lipid peroxides in a way similar to its reaction with H2O2 and produce alkoxy (R ‐ O·) and peroxy radicals (R ‐ O2 ·). The resulting peroxy radical leads to propagation of lipid peroxidation [57].
Iron can also react with lipid peroxides in a way similar to its reaction with H2O2 and produce alkoxy (R ‐ O·) and peroxy radicals (R ‐ O2 ·). The resulting peroxy radical leads to propagation of lipid peroxidation [57].
Lipid peroxidation can proceed until the lipid radicals interact with one another and/or a “chain breaker,” such as vitamin E, forming a no-radical species.
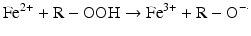
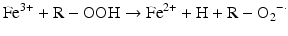
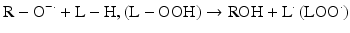 Iron compounds such as free Hb can also catalyze peroxidation of purified arachidonic acid and other polyunsaturated fatty acids within normal cell membranes in the presence of H2O2 and •O2− [58].
Iron compounds such as free Hb can also catalyze peroxidation of purified arachidonic acid and other polyunsaturated fatty acids within normal cell membranes in the presence of H2O2 and •O2− [58].
The CNS is separated from the systemic circulation by the blood–brain barrier (BBB), a tight epithelial barrier analogous to that of the mammalian duodenum, which is the site of absorption of dietary iron. After absorption, iron in its oxidized (ferric) form binds to serum transferrin and is distributed throughout the general circulation. The presence of a BBB explains the relative independence of the brain iron from the total body iron content [59].
Incorporation and Transport of Iron in the Nervous System
Iron is transported to the brain via the BBB which is composed of endothelial cells in small vessels throughout the brain that contain tight junctions which regulate the brain iron levels.
Iron incorporation and transport in the brain depends on interactions between the endothelial cells and astrocytes. Brain endothelial cells express the transferrin receptor 1 (Tf R1) in their luminal membrane. This receptor binds iron-loaded transferrin and internalizes this complex in endosomes.
Within the endosomes, the acid environment facilitates the release of ferric iron from transferrin and this is followed by reduction of ferric to ferrous iron by action of endosomal reductases. The mechanism by which iron is released from transferrin is transported from the interior of the brain endothelial cells to the interstitial fluid is a matter of controversy [60, 61].
One possibility is that ferrous iron is transported from the endosome to the cytosol by the divalent metal transporter-1 (DMT1) and then exported into the extracellular fluid by action of ferroportin [60].
However, there is some disagreement as to the degree of expression of DMT1 and ferroportin and their contribution to iron transport across the brain endothelial cells. Alternatively, it has been proposed that the transferrin-Tf R1 receptor complex is transported within the endosomes from the luminal to the abluminal surface where endosomes release iron at the interface between the endothelial cells and astrocyte end-foot processes [61].
The end-foot processes express ceruloplasmin, which acts as a ferroxidase that oxidizes newly released ferrous iron to ferric iron and binds to the transferrin in the brain interstitial fluid [60–62]. Transferrin is the main source of iron for neurons, which express high levels of Tf R1. Whereas transferrin is synthesized by oligodendrocytes, the primary source of transferrin in the brain interstitium is its diffusion from the ventricles.
It has been shown that transferrin uptake and the ratio of iron to transferrin uptake by the brain decrease with age, and the transferrin recycling time increases with age [63].
Neuronal Iron Homeostasis
There is a tight regulation of the cytosolic iron pool in brain cells, and is critical for two reasons: (1) iron is an important source for numerous cytosolic, mitochondrial, and nuclear ferroproteins; and (2) excessive accumulation of free cytosolic ferrous iron predisposes to oxidative stress and cytotoxicity.
Iron regulatory proteins sense cytosolic iron levels and interact with iron-responsive elements and regulate translation of mRNA encoding for proteins involved in iron uptake, storage, and mobilization, including Tf R1, ferritin, and ferroportin.
In the cytosol, the storage protein ferritin sequesters and reduces levels of free iron. Ferritin consists of a heavy [ferritin heavy chain (FTH1)] subunit that catalyzes the rapid oxidation of ferrous to ferric iron and a light (FTL) sub-unit that may be involved in the nucleation of the iron core within the protein shell. Thus, ferritin has a dual function of iron detoxification and iron reserve [64].
Ferritin is also present in axons and may transport iron to the synapse. Ferroportin, present in synaptic vesicles, may allow release of ferrous iron at the synapses [60, 61]. Neuromelanin is an insoluble pigment produced from oxidation of excess cytosolic catechols and is present in granules in dopaminergic neurons of the substantia nigra and in noradrenergic neurons of the locus ceruleus. Neuromelanin binds iron avidly forming stable complexes and sequesters large amounts of iron in those cells [65].
Mitochondrial iron is required for heme biosynthesis and for the generation of iron/sulfur (Fe/S) clusters in many essential enzymes. Iron is transported into the mitochondria by mitoferrin, a transporter expressed in the inner mitochondrial membrane.
Frataxin is a mitochondrial iron chaperone that is involved in the biosynthesis of Fe/S clusters by interactions with critical assembly proteins. [62, 66–68] The Fe/S clusters form the prosthetic group of many enzymes of the respiratory chain and also constitute the main mechanism for iron exit from the mitochondria.
The major route of iron export out of the brain is via the cerebral spinal fluid (CSF) and its reabsorption into the blood from the subarachnoid space. The concentration of transferrin in CSF is very low and its capacity to export iron is limited. Lactoferrin, ferritin, and non-protein-bound iron are also present in the CSF and may contribute to iron export. In normal conditions, iron concentration is very low but can increase considerably under pathologic circumstances as discussed below. Microglia and other phagocytic cells are additional important mediators of iron export after cell death and intracerebral hemorrhage.
Iron accumulates in the brain as a function of age, primarily in the form of ferritin, particularly in the microglia and astrocytes, but also in the oligodendrocytes. The brain areas richest in iron are the globus pallidus, substantia nigra, putamen, caudate nucleus, dentate nucleus, and frontal cortex [69].
Impaired regulation of iron homeostasis in those cells may lead to either excessive accumulation of free cytosolic iron or decreased iron availability for critical enzymes. Free cytosolic ferrous iron reacts with endogenously generated hydrogen peroxide to yield hydroxyl radicals, which damage cell membranes. A recent study identified a signaling cascade that links the activation of the N-Methyl-D-aspartate (NMDA) receptors, which mediate glutamate triggered excitotoxicity, with iron homeostasis. This cascade involves activation of nitric oxide synthase and adaptor proteins that interact with ferroportin [70].
Iron deposition or dysregulation occurs in several neurodegenerative disorders, including sporadic AD, PD, FA, and NBIA.
Brain Neurodegenerative Disorders and Iron
Dysregulation of iron homeostasis is also a critical feature of AD, PD, NBIA and FA. Although these diseases have their own distinctive features, they all have one thing in common: accumulating iron in the brain. Increased iron in the brain, rich in oxygen and fatty acids, provides an ideal environment for oxidative stress and possible irreparable tissue damage.
The link between iron and neurodegenerative disease provides potential therapeutic targets for these disorders. If, indeed, iron and/or oxidative processes are involved in the pathogenesis of neurodegenerative disorders, approaches such as iron chelation therapy and antioxidant supplements might help to slow the degenerative processes or ameliorate brain tissue injury.
Alzheimer Disease
AD is a progressive degenerative disease with a gradual deterioration in memory, cognition, behavior, and the ability to perform activities of daily living. Evidence of increased brain metal levels such as iron and copper has been associated with AD [71].
The amyloid-beta (Aβ) plaques in the brain are the hallmark pathologic features of AD and are derived from the cleavage of amyloid precursor protein (APP). Deposition of fibrillar aggregates of Aβ in the brain parenchyma, which is caused by Aβ overproduction, impaired clearance, or both, has been hypothesized to explain the cause of AD [72].
APP has binding sites in its amino-terminal domain and in the Aβ domain for copper and iron. Iron is primarily complexed with ferritin and concentrated in the neuritic processes associated with amyloid plaques [69, 73]. Iron might have a direct impact on plaque formation through its effects on APP processing by alfa-secretase, deposition of Aβ, and oxidative stress [63, 73].
High levels of iron may interact with the Aβ peptide, leading to the reduction of molecular oxygen to superoxide and eventually to H2O2 by reducing iron. It has also been demonstrated that overexpression of the carboxyl terminal fragment of APP (Aβ) significantly reduces the level of copper and iron in the transgenic mouse brain. This suggests a role for APP and Aβ in physiologic metal regulation in AD [74].
Additionally, overexpression of melanotransferrin has been reported in AD. Because purified melanotransferrin can bind iron, it has been proposed as another protein that also might be involved in iron transportation [75].
The role of iron in the pathogenesis of AD is thought to be related to enhance oxidative stress mediated by free iron. Transferrin, ferritin, and iron regulatory protein 2 also have been associated with neurodegeneration in AD. The latter might be responsible for the disturbance in brain iron homeostasis and the overall decompartmentalization of iron and the resulting oxidative processes suggestive of AD [74].
Genetic alterations specific to iron-management proteins, including HEF1 mutations (associated with congenital hemochromatosis) or the transferrin subtype C2, may increase the risk of AD [76].
Friedreich Ataxia
FA is an autosomal recessive neurodegenerative disease characterized by degenerative atrophy of the posterior columns of the spinal cord followed by the spinocerebellar tracts and corticospinal motor tracts, leading to progressive ataxia, sensory loss, and muscle weakness. It is also characterized by degeneration of large sensory neurons of the dorsal root ganglion, cerebellum (particularly the iron-rich dentate nucleus), and cardiomyocytes.
FA is an autosomal recessive disorder resulting from a large guanine adenine adenine (GAA) triplet-repeat expansion in the first intron of the Friedreich ataxia (FRDA) gene, resulting in a reduction in expression of the encoded protein, frataxin [66–68, 77].
Frataxin is a mitochondrial protein and is suggested to have a role in mitochondrial iron transport or in iron-sulfur assembly and transport. High levels of iron in the mitochondria can react with superoxide (•O2−) and H2O2 to produce the hydroxyl radical (•OH), which can oxidize cellular components, damage respiratory chain complexes, and result in cellular injury and eventually cell death.
Whereas excess mitochondrial iron is detected in neurons and cardiomyocytes from affected patients, indicating a predisposition to oxidative stress, there is evidence that impairment of heme and Fe/S cluster biosynthesis is the most likely proximate cause of neurodegeneration in this disorder [62, 66–68, 77].
Parkinson Disease
PD is a progressive disorder manifesting as tremor at rest, bradykinesia, gait abnormalities, rigidity, postural dysfunction, and loss of balance. Iron has been suggested to be responsible for nigrostriatal dopamine neuron degeneration in PD owing to its ability to produce toxic ROS and cause lipid peroxidation [81].
PD is characterized by iron accumulation in dopaminergic neurons of the substantia nigra. Free cytosolic iron may trigger oxidative stress and promote alfa-synuclein aggregation with deposition of Lewy bodies [69].
Lewy bodies can have deleterious effects on the extrapyramidal system and on psychomotor function [82, 83]. The presence of the pigment neuromelanin in the substantia nigra in PD also might result in iron accumulation because neuromelanin can function like ferritin and store iron [84].
Over expression of lactoferrin (a protein that reversibly binds iron) receptors on neurons and microvessels in regions of neuronal degeneration in PD-affected brain tissue suggests a possible link to iron overload in affected brain regions [82].
All these mechanisms suggest that disturbances in iron homeostasis and metabolism in PD occur at several levels, such as iron uptake, storage, intracellular metabolism, release, and posttranscriptional control [85].
As indicated in the preceding text, a disturbance in iron homeostasis can provide a favorable condition in which free iron, via generation of ROS, causes permanent tissue damage. Neuromelanin may exert neuroprotective action at early stages of PD because it prevents free iron accumulation and thus hydroxyl radical production and formation of neurotoxic dopamine quinones [65].
However, in advanced stages, extravasation of neuromelanin granules from dying neurons may attract and activate microglia, causing release of neurotoxic molecules leading to cell injury [65]. Both iron chelation and over expression of iron-sequestering ferritin have been shown to be protective in animal models of PD [69].
Type I Neurodegeneration with Brain Iron Accumulation
Type I NBIA (NBIA-1) was formerly known as Hallervorden-Spatz syndrome or pantothenate kinase-associated neurodegeneration.
NBIA-1 is a rare, genetically determined neurodegenerative disorder characterized by extrapyramidal dysfunction and mental deterioration. Iron accumulates mainly in the globus pallidus and the pars reticularis of the substantia nigra, and presents as brown-pigmented iron deposits [86].
NBIA comprises a clinically and genetically heterogeneous group of disorders that include pantothenate kinase–associated neurodegeneration (PKAN), infantile neuroaxonal dystrophy, neuroferritinopathy, and hereditary aceruloplasminemia [87]. These disorders are characterized by iron accumulation and cell loss affecting primarily the globus pallidus and, in many cases, retinal photoreceptors [87].
PKAN is caused by a mutation of the gene encoding pantothenate kinase 2 (PANK2), the key enzyme in the synthesis of mitochondrial coenzyme A. The mutation in a novel pantothenate kinase gene, PANK2, is predicted to cause the accumulation of cysteine, which binds iron and causes oxidative stress in the iron-rich globus pallidus [88].
Neuroferritinopathy is a dominantly inherited, adult-onset disorder caused by mutations in the ferritin light chain (FTL1) gene [89–91]. It results in accumulation of ferritin-iron aggregates in neurons and glial cells of the globus pallidus, substantia nigra, striatum, and cerebellum. Vacuolated glial and neuronal nuclei may be characteristic of the disease [90]. Focal onset limb dystonia or chorea, orolingual dyskinesia, dysarthria, aphonia, and dysphagia are prominent features.
Hereditary aceruloplasminemia is a rare autosomal recessive disorder caused by a mutation of the ceruloplasmin gene, leading to iron overload in the brain and reticuloendothelial system. It manifests with anemia, diabetes, retinal degeneration, and progressive neurologic disorder [92, 93]. A characteristic neuropathologic finding is the presence of enlarged deformed astrocytes and accumulation of spheroid-like, grumose foamy deposits in the astrocytic foot processes [94].
Application of high-resolution imaging technologies such as electron energy-loss spectroscopy and electron tomography may allow early identification of the cell type and intracellular location of iron deposits in patients with neurodegenerative disorders.
If, indeed, iron and/or oxidative processes are involved in the pathogenesis of neurodegenerative disorders, approaches such as iron chelation therapy and antioxidant supplements might help to slow the degenerative processes or to ameliorate brain tissue injury. Several chelators are currently under investigation for treatment of these and other disorders associated with abnormal brain iron homeostasis.
The neuroprotection that is provided by iron chelators in animal models indicates that iron-chelation therapy could be a viable neuroprotective approach for treatment of disorders such as PD or AD.
Manganese
Manganese is an essential nutrient that is common in the environment. It is the fifth most abundant metal and the twelfth most abundant trace element in the earth’s crust [95]. It is necessary for the adequate functioning of the human CNS, but it also has the potential to produce neurotoxic effects when, depending on the route and dose of exposure, it accumulates in an organism (particularly in the brain) exceeding the homeostatic range [96]. It is released into the environment as a product of industrial activities, the use of the manganese-containing pesticides such as Maneb® and Mancozed®, and through the use of methyl-cyclopentadienyl manganese tricarbonyl, as a gasoline antiknock agent (Agency for Toxic Substances and Disease Registry, ATSDR, 2000).
The vast majority of studies on neurotoxic effects of manganese were conducted in occupational settings where exposure occurs mainly through inhalation of airborne particulates (ferroalloy smelting, welding, mining, battery assembly, etc.). Several studies have demonstrated the impact of manganese toxicity in adults, resulting in cognitive, neurological, motor, and psychological impairment [97–102].
In the past years, many studies have investigated possible overexposure of children to manganese and subsequently the neuropsychological effects produced. It is generally accepted that children are at greater risk than adults exposed to the same contaminants from the environment [103].
In general, with normal dietary consumption, systemic homeostasis of manganese is maintained. Although very low levels of manganese in air, soil, water, and food are normal, nevertheless, excess exposure can occur by inhalation in areas where there is high manganese concentration in air and dust, or by drinking water that has had long contact with bedrock enriched in manganese, or by consuming high amounts of food sources rich in the trace element. The highest concentrations are found in nuts, legumes, and blueberries teas [104]. Other authors have hypothesized the possibility of overexposure to manganese through ingestion of infant milk formula [105], or even by iatrogenic manganese exposure than occurs in individuals receiving total parenteral nutrition resulting in increased concentrations of manganese in the brain [106]. Furthermore, exposure to at-risk populations with compromised or immature BBBs or underdeveloped excretory pathways (i.e., children) can also result in increased brain manganese levels [107].
Several factors could predispose children to manganese overexposure and subsequent toxic effects. Exposure to manganese by ingestion or inhalation can have different consequences in children than in adults and through different mechanisms [108]. First, children are exposed to a larger amount of manganese from inhalation because of their higher breathing rates (the ratio of inhaled air/weight is much higher in children because of the lower body mass), and greater intestinal absorption rate [103, 109]. Absorption can be as high as 80 % in neonates compared with 1–5 % in adults [11]. Second, high demand for iron linked to growth could further enhance the absorption of ingested manganese [110]. Third, a low excretion rate was observed in infants because of their poorly developed biliary excretion mechanism [111]. In fact, exposure during this period may result in increased delivery of manganese to the brain and other tissues.
Neurotoxic effects resulting from excessive manganese exposure were first described in 1837 by Couper in Scottish laborers who were grinding manganese black oxide in the chemical industry [112]. Neurological symptoms of manganism include decreased memory and concentration, fatigue, headache, vertigo, equilibrium loss, insomnia, tinnitus, trembling of fingers, muscle cramps, rigidity, alteration of libido, and sweating [113]. Many reports of neurotoxic effects in manganese-exposed workers were later published [100], and the definition of manganese intoxication has evolved to include subclinical signs of intoxication indicated by alterations of neurobehavioral functions [114].
Manganese plays a role in immune response, blood sugar homeostasis, ATP regulation, reproduction, digestion, and bone growth [115]. It is a necessary component of metalloenzymes such as manganese superoxide dismutase, arginase, phosphoenol–pyruvate decarboxylase, and glutamine synthetase. This glutamine synthetase enzymeconverts glutamate into glutamine. Glutamine synthetase [116].
Manganese shares several characteristics with iron; both are transition metals with valences of 2+ and 3+ in physiological conditions and proximate ionic radius. In addition, as manganese and iron both strongly bind to transferrin and accumulate in the mitochondria, low iron stores are associated with increased manganese uptake and retention in the blood [117], and increment of the accumulation in CNS. More than 2,000 million people on our pleanet, mainly children and pregnant women (and/or in fertile age), manifest ferropenic anemia after inadequate iron absorption. The potential effects associated to the accumulation in CNS of manganese in these populations represent a sanitary challenge of great magnitude. Manganese can accumulate in the CNS, particularly the basal ganglia but also the cortex. Exposure to manganese has been shown to interfere with several neurotransmitter systems, especially in the dopaminergic system in areas of the brain responsible for motor coordination, attention, and cognition [117, 118]. Manganese is a potent dopamine oxidant, which could explain the toxic lesions in certain dopaminergic brain regions [119]. Excessive exposure could result in dopamine receptor loss or inactivation through damage to the membrane mediated by free radicals or cytotoxic quinones generated by the manganese catalyzing effect on autooxidation of this neurotransmitter [120]. The correlation between manganese and hyperactive behavior is probably a result of the dopaminergic and gamma-aminobutyric acidergic systems, which play a role in hyperactivity in children [121, 122].
One hypothesis for the toxic mechanism of manganese is the production of excess free radicals in the nerve cell, potentiating lipid peroxidation, and resulting in tissue destruction [122, 123]. Manganese neurotoxicity has been extensively studied and a lot has been learned about its mechanism of action at the cellular and molecular levels and the detection of subclinical effects at low exposures. In the last few years, several literature reviews have been published on aspects such as neurotoxic effects on exposed laborers [118, 124], the application of magnetic resonance imaging [125], neuropsychological testing for the assessment of manganese neurotoxicity, manganese neurotoxicity focused on neonates [126], neurotoxicology of chronic manganese exposure in nonhuman primates [127], and manganese exposure and neuropsychological effect on children and adolescents [108, 128].
In most studies, authors observed that manganese exposure was associated with poorer cognitive functions and hyperactive behavior. Many have suggested that manganese exposure is related to cognitive, motor, and behavior deficits in children. Some of them found an adverse effect of manganese on cognitive function, and overall an inverse association between manganese exposure and IQ [129–131]. Others studies have focused on motor effects of manganese, finding a positive association. Very few studies focused on the effects of manganese on children’s motor skills, although data on motor effects in adults, occupationally or environmentally exposed to manganese, have been reported [132, 133].
Related posts:
 Knowledge, by Philosophy or Science? Psychotherapy or Neuroscience?
Neurovascular Cognitive Alterations: Implication of Brain Renin–Angiotensin System
Animal Models of Psychopathology and Its Relation to Clinical Practice
Adenosine in the Neurobiology of Schizophrenia: Potential Adenosine Receptor-Based Pharmacotherapy
Translating the Glutamatergic Hypothesis of Schizophrenia Through Homeostatic Regulation of Brain Glycine
Mindfulness and Neuroimaging
Knowledge, by Philosophy or Science? Psychotherapy or Neuroscience?
Neurovascular Cognitive Alterations: Implication of Brain Renin–Angiotensin System
Animal Models of Psychopathology and Its Relation to Clinical Practice
Adenosine in the Neurobiology of Schizophrenia: Potential Adenosine Receptor-Based Pharmacotherapy
Translating the Glutamatergic Hypothesis of Schizophrenia Through Homeostatic Regulation of Brain Glycine
Mindfulness and Neuroimaging
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


