Fig. 20.1
Diagrammatic representation of a classic pain pathway. In this simplified representation only the commonest of noxious stimuli have been included (chemically-triggered irritation, burning and mechanical damage)
Remarkably, the nociceptor is also subjected to influences emerging from their innervations targets, nerves and also the spinal cord. These extrinsic signals contribute to the function and phenotype of the nociceptor and are added to the intrinsic properties of the nociceptor itself.
That nociceptors and the ability to sense pain are central to survival in normal individuals can be illustrated by the unfortunate patients carrying a mutation in TrkA, the receptor for nerve growth factor (NGF). These patients suffer from hereditary sensory and autonomic neuropathy type 4, in which because of the lack of a functional TrkA, nociceptors failed to survive [9]. This condition produces congenital pain hyposensitivity, causing the patients to burn and chew their tongues and lips, and as a result of undetected damage, lose the tips of their fingers and damage their joints. Clearly, ignorance of noxious stimuli is not bliss: this is the Yin side of pain, a necessary mechanism set to protect us from inflicting self-damage (either by action or omission) or a warning signal to alert us that something in our body is not right. Other examples in which there is an innate inability to sense pain not associated to nociceptor loss but rather to a lack of key molecular mediators of pain in these neurons, are those carrying mutations in voltage-dependent Na+ channels such as SCN9A (the gene encoding the alpha subunit of Nav1.7 voltage-gated sodium channel) [10, 11] as well as SCN11A (which encodes for Nav1.9) [12–14].
Finally, we must bear in mind that, beyond the events occurring at the cellular and molecular level, pain is a phenomenon that has a physical and a psychological dimension. In the specific case of chronic or maladaptive or pathological pain, these two aspects are characterized by the occurrence of vicious circles (understood as self-sustaining and self-preserving mechanisms that reinforce undesirable/uncomfortable behaviors).
The psychological vicious cycle begins with feelings of anger, anxiety, fear, etc. arising from the presence of pain (particularly if it is chronic, moderate to severe in intensity or if it is spontaneous and unpredictable). These feelings drive the individual to a bad, poor mood, which if prolonged in time, could lead to depression. In turn, depressive syndromes can accentuate the subjective side of pain, leading to increased pain perception (even when the intensity of the pain remains unchanged over time). This takes us back to the beginning and the cycle is then perpetuated unless the pain is effectively suppressed.
The physical vicious cycle typically begins when the person avoids doing physical activity because of his/her pain. The longer this avoidance goes on, the more deconditioning occurs. The lack of activity has several implications: the patient becomes less active, hence with lesser social life and growing isolation—this feeds back to the poor mood and the depression and also leads to further activity avoidance. Again, the longer this state lasts, the more difficult it becomes to return to physical activity, which nurtures a heightened level of physical discomfort and eventually causes more pain. This cycle is then closed, and links to the psychological one (see Fig. 20.2). That type of pathological pain is seen as the Yang, the one that no longer serves the protective role of the warning system, and instead becomes a debilitating and hard to treat medical condition with significant clinical relevance [15, 16].
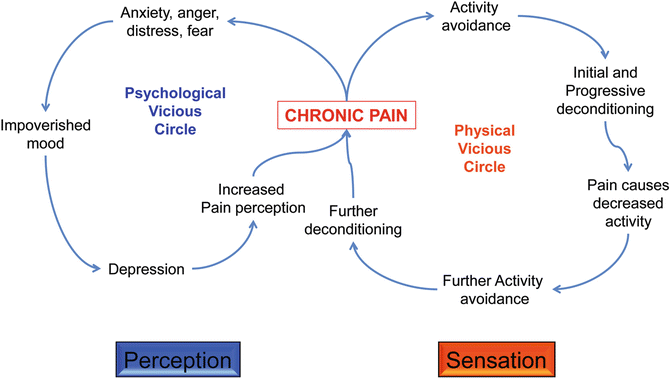
Fig. 20.2
The vicious circles of chronic pain. The flow diagram illustrates the likely sequence of events leading to depression and avoidance of physical activity, two of the most important side effects of chronic pain with a significant impact on patient’s quality of life
In summary, in this chapter we briefly present the main aspects of pain. First we present evidence on how nociceptors (as the mediators of pain) acquire their specialized molecular phenotype. Second, we comment on how they transduce noxious stimuli and transfer input to the CNS. Finally, we elaborate on how some of the adaptive and maladaptive functional and phenotypic changes that occur in them in conditions of inflammation and illness lead to spontaneous pain and pain hypersensitivity.
The Terminology of Pain
There are a number of key concepts in the field of pain and pain research that we need to define as best as we can so as to properly understand the extent of the problem and also to put into context many of the challenges that physicians (and other health professionals) face during diagnosis and treatment of pathological pain.
According to the Online Etymology Dictionary, the word pain probably originated from the Latin poena, meaning “punishment, penalty, retribution, indemnification” (in Late Latin also “torment, hardship, suffering”) and from the Greek poine, that is “retribution, penalty, quit-money for spilled blood” and also possibly from PIE*kwei- “to pay, atone, compensate”. The earliest sense in English survives in the phrase on pain of death. The word pain also has a root in the Old French (eleventh century) peine “difficulty, woe, suffering, punishment, Hell’s torments”. Pains as in “great care taken (for some purpose)” is first recorded in the 1520s (in the singular in this sense, it is attested from c.1300). The first record of the term pain-killer dates from 1853. These concepts of pain as being essentially associated to the idea of hardship and punishment is in agreement with its endurance bringing spiritual elevation and purification, ideas that were predominant in Medieval and Modern times.
Contemporaneously, the International Association for the Study of Pain (IASP) set a permanent committee in charge of periodically reviewing the definitions for a number of key terms. Here we pursue those definitions, which are presented as listed in the IASP website (http://www.iasp-pain.org/AM/Template.cfm?Section=Pain_Definitions) with some additional comments where it is pertinent to make clarifications or where debate is ongoing about the exact meaning of a term.
Pain: An unpleasant sensory and emotional experience associated with actual or potential tissue damage, or described in terms of such damage.
First must note that pain is always subjective. Therefore, individuals report their perception as being painful, and they do so verbally. However, the inability to communicate verbally does not negate the possibility that an individual is experiencing pain and is in need of appropriate pain-relieving treatment.
We learn about the meaning of the word “pain” through experiences in early life. The episodes linked to the use of the term are normally related to injury and possible tissue damage. Accordingly, pain is that experience we associate with actual or potential tissue damage. It must be noted that although pain is unquestionably a sensation in a part or parts of the body, it is also always unpleasant and therefore also an emotional experience. In line with this argument, experiences which resemble pain but are not unpleasant, e.g., pricking, should not be called pain.
Unpleasant abnormal experiences (called dysesthesias and defined below) may also be called pain but are not necessarily so because, subjectively, they may not have the usual sensory qualities of pain. Many people report pain in the absence of tissue damage or any likely patho-physiological cause; usually this is attributable to psychological rather than physiological reasons. Unfortunately, there is usually no way to distinguish their experience from that due to actual tissue damage if we limit our investigations to the subjective report. The position of the IASP Committee is that if the patients regard their experience as pain, and if they report it in the same ways as pain caused by tissue damage, it should be accepted as pain. This definition clearly avoids tying pain to the stimulus causing it. Activity induced in nociceptors and nociceptive pathways by a noxious stimulus is not considered pain, which is always a psychological state.
Neuropathic pain : Pain caused by a lesion or disease of the somatosensory nervous system.
We must allude to the fact that neuropathic pain is a clinical description (and not a diagnosis) which requires the presence of a demonstrable lesion or a disease that satisfies the established neurological diagnostic criteria. Lesion is commonly used when diagnostic investigations (e.g., imaging, neurophysiology, biopsies, laboratory tests) reveal an abnormality or when there was obvious trauma. The term disease is typically used when the underlying cause of the lesion is known (e.g., stroke, vasculitis, diabetes mellitus, genetic abnormality). Somatosensory refers to information about the body per se including visceral organs, rather than information about the external world (e.g., vision, hearing, or olfaction). The presence of symptoms or signs (i.e., touch-evoked pain) alone does not justify the use of the term neuropathic. Some diseases, such as trigeminal neuralgia, are currently defined by their clinical presentation rather than by objective diagnostic testing. Other diagnoses such as post-herpetic neuralgia are normally based on the clinical history of the patient. It is common when investigating possible neuropathic pain that diagnostic testing yield inconclusive or even inconsistent data. In such instances, clinical judgment is required to reduce the totality of findings in a patient into one putative diagnosis or concise group of diagnoses.
The main difference between central neuropathic pain and peripheral neuropathic pain is that the former is caused by a lesion or disease of the central somatosensory nervous system, while the latter is caused by a lesion or disease of the peripheral somatosensory nervous system.
In sharp contrast to neuropathic pain, nociceptive pain is pain that arises from actual or threatened damage to non-neural tissue and is a result of the activation of nociceptors. In fact, this term is used to describe pain occurring with a normally functioning somatosensory nervous system to contrast with the abnormal function seen in neuropathic pain. In other words, the protective, normally arising acute pain caused by a sudden injury for example, is called nociceptive while pain arising from an underlying malfunction or damage in the somatosensory system is termed as neuropathic (provided the damage has been diagnosed and established as the cause for the reported pain, usually chronic in duration). A patient may occasionally exhibit a combination of symptoms which in turn can be described as being partly neuropathic and partly nociceptive. Figure 20.3 summarizes what we have described and provides a few examples of each type of pain.
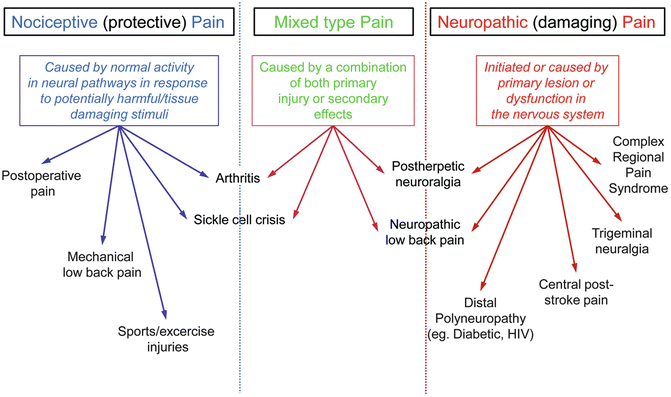
Fig. 20.3
Comparative definitions of nociceptive, neuropathic and mixed pain with examples of clinically relevant conditions typically classed into each type
When the pain is located in the distribution of a single nerve or nerves, it is referred to as neuralgia, while a demonstrable inflammation of a nerve or nerves is called neuritis. Note that pain associated with inflammation of tissues other than nerves could be termed inflammatory pain. However, this may lead to confusion because chronic inflammation resulting from conditions such as arthritis often causes pain syndromes that fit well within the definition of neuropathic pain. Furthermore, many clinical conditions that present with chronic pain are associated with ongoing chronic inflammation, which is believed to play a role in both, causing and maintaining this type of pathological pain.
Neuropathy is a disturbance of function or pathological change in a nerve: in one nerve, mononeuropathy; in several nerves, mononeuropathy multiplex; if diffuse and bilateral, polyneuropathy. Note that neuritis is a special case of neuropathy.
Patients occasionally present with a combination of symptoms which constitute a syndrome. Such is the case of causalgia, a syndrome of sustained burning pain, allodynia, and hyperpathia after a traumatic nerve lesion, often combined with vasomotor and sudomotor dysfunction and later trophic changes. Hyperpathia, on the other hand, is a painful syndrome characterized by an abnormally painful reaction to a stimulus, particularly a repetitive stimulus, presenting with an increased threshold and the pain is often explosive in character.
Additionally, there are signs or symptoms that are associated with the different types of clinically relevant pain and need to be defined. They include allodynia, hyperalgesia, dysesthesia, hyperesthesia, hypoalgesia and paresthesia. Next, we briefly introduce each term, followed by its accepted IASP definition.
Allodynia is pain resulting from a stimulus that does not normally provoke pain. In this case, a stimulus that normally does not cause pain, leads to an unexpectedly painful response. This is a clinical term that does not imply a mechanism. Allodynia may be observed following application of different types of somatosensory stimuli to various other tissues. Allo means “other” in Greek and is a common prefix for medical conditions that diverge from the expected. Odynia is derived from the Greek word “odune” or “odyne” meaning “pain” which is used in “pleurodynia” and “coccydynia” and is similar in meaning to the root from which we derive words with -algia or -algesia in them.
The term allodynia was originally introduced to separate hyperalgesia from hyperesthesia, the conditions seen in patients with lesions of the nervous system where touch, light pressure, or moderate cold or warmth evoke pain when applied to apparently normal skin.
There are a number of potential problems with the definition of allodynia. First, how can we be certain that a strong pinch applied to the skin of a normal individual does not cause significant tissue damage, even in the short term? Can we consider that sensitized skin (e.g., sunburn) constitutes a sort of peripheral pain amplifier that can result in light tactile stimulation leading to pain? And yet, sunburned skin is technically normal skin that has transiently been affected by excessive solar irradiation that is unlikely to result in any permanent damage. These complicating factors are By “implicated” we mean that the definition of allodynia implies an abnormal pain processing being associated to abnormal tissue (or damaged tissue) whereas in the example of sunburn skin, the skin is physiologically normal.
It is also important to recognize that allodynia involves a change in the quality of a sensation, whether tactile, thermal, or any other sort. The original modality is normally non-painful, but the response it triggers is painful. Thus there is a loss of specificity of a sensory modality. By contrast, hyperalgesia (see later) represents an augmented response in a specific mode, that is, pain. In allodynia, the stimulus mode and the response mode differ, unlike the position with hyperalgesia.
Hyperalgesia is increased pain from a stimulus that normally provokes pain; in other words, hyperalgesia reflects increased pain on suprathreshold stimulation. As with allodynia, this is a clinical term that does not have any mechanistic implications (i.e., it does not convey information about the actual ontogeny of the phenomenon being described by the term). For pain evoked by stimuli that usually is not painful, the term allodynia is preferred (see above), while hyperalgesia is more appropriately used for cases with an increased response at a normal threshold, or at an increased threshold (e.g., in patients with neuropathy). It should also be recognized that with hyperalgesia the stimuli and the response are both in the same sensory mode. Current evidence suggests that hyperalgesia is a consequence of perturbation of the nociceptive system with concurrent peripheral or central sensitization, or both. However, it is important to distinguish between the clinical phenomena, which this definition emphasizes, as well as the interpretation, which may well change as knowledge advances.
Hyperesthesia is an increased sensitivity to stimulation, excluding the special senses. Hyperesthesia can refer to various modes of cutaneous sensibility including touch and thermal sensation without pain, as well as to pain. The word is used to indicate both diminished threshold to any stimulus and an increased response to stimuli that are normally recognized. In this sense, hyperesthesia includes both allodynia and hyperalgesia, but the more specific terms should be used wherever they are applicable.
Dysesthesia is an unpleasant abnormal sensation (often painful) while paresthesia is just an abnormal sensation (e.g., tingling under the skin), in both cases regardless of whether these sensations are spontaneous or evoked. From these definitions it is clear that hyperalgesia and allodynia are both special cases of dysesthesia. It may also be true that dysesthesia is a particular form of paresthesia, however,the reverse is not true.
Hypoalgesia is a diminished pain in response to a normally painful stimulus. This term refers only to the occurrence of relatively less pain in response to stimulation that normally causes pain. On the other hand, hypoesthesia refers to the case of diminished sensitivity to stimulation that is normally painful.
Finally, analgesia is understood to be absence of pain in response to stimulation which would normally be painful. As with allodynia, the stimulus is defined by its usual subjective effects.
Some aspects of pain are relevant to the understanding of the mechanisms underlying its occurrence. They suggest what physiological parameters are likely to be affected at the cellular level, and are therefore, useful to guide research efforts to what is causing pain. In this category, we encounter concepts such as pain threshold, defined as the minimum intensity of a stimulus that is perceived as painful. Although this definition is accurate, in practice the threshold itself is really the experience of the patient, whereas the intensity measured is an external event. It has been a common mistake for many pain researchers to define the threshold in terms of the stimulus, which should be avoided. Nonetheless, the threshold stimulus can be recognized as such and measured. In psychophysics for example, thresholds are defined as the level at which 50 % of stimuli are recognized. In that case, the pain threshold would be the level at which 50 % of stimuli would be recognized as painful. We should keep in mind that the stimulus is not pain and it cannot be a measure of pain. Other subjective experience of the individual is the pain tolerance level, that is, the maximum intensity of a pain-producing stimulus that a subject is willing to accept in a given situation. As with the pain threshold, it is not a measure of pain. However, both measurements are important because lowered pain thresholds or tolerance levels suggest changes in the excitability of the nociceptors, which are likely to involve, for example, alterations in the electrical properties of the neuronal membrane. Hence their importance as tools to gain insights into the mechanisms underlying pain must be pointed out.
There has been extensive research into the ability of nociceptors to exhibit increased responsiveness to their normal input, and/or recruitment of a response to normally sub-threshold inputs. This complex phenomenon is called sensitization and can include a lowered threshold and an increase in supra-threshold responses. Spontaneous discharges and increases in receptive field size can also occur. This is a neurophysiological term that can only be applied when both input and output of the neural system under study are known (e.g., by controlling the stimulus and measuring the neural event). Clinically, sensitization can only be inferred indirectly from phenomena such as hyperalgesia or allodynia. It has been shown that these sensations involve a degree of increased nociceptive responsiveness to external stimuli. If the sensitization affects the function of central neurons only while peripheral neurons function normally, we refer to central sensitization. When what we observe is an increased responsiveness and reduced threshold of nociceptive neurons in the periphery to the stimulation of their receptive fields, we are observing peripheral sensitization.
Up to this point we have used terms such as “nociceptor” or “nociceptive” without properly defining them. For that matter we have not as yet clearly stated what we understand by the term nociception . The simplest possible definition states that nociception is the neural process of encoding noxious stimuli. Note, however, that this definition does not necessarily imply pain sensation. As a matter of fact, consequences of encoding may be autonomic (e.g., elevated blood pressure) or behavioral (e.g., motor withdrawal reflex or more complex nocifensive behavior). The former is not usually linked to pain per se. Having said this, we need to establish the difference between a nociceptive neuron and a nociceptor. In general, a nociceptive neuron is a central or peripheral neuron in the somatosensory nervous system that is capable of encoding noxious stimuli. Notice that this definition does not explicitly state any specific physiological properties for these neurons, and therefore can be applied to a number of neuronal types not necessarily linked to pain sensation. This is not true for nociceptors, whose function is to encode and transduce noxious stimuli, and to do so normally in response to high stimulation thresholds.
The Nociceptor
In all clinically relevant situations, and at the core of pain sensation, lies the specialized neuron we call nociceptor (reviewed in detail by [17]). As excitable cells with receptive fields projecting to the external as well as the internal environment of the organism, they possess a large number of receptors acting as sensors. In turn, the activation of these receptors leads to signalling events terminating in a number of possible targets: the nucleus, where they promote synthesis of new proteins; the proteins of the cell membrane involved in controlling neuronal excitability, e.g., ion channels or even other receptors, also membrane bound, that can be sensitized to react to the presence of small quantities of certain chemicals; and so on. We must add to this cellular complexity that a given subtype of nociceptor (e.g., mechanoceptive, thermoceptive, polymodal, etc.) carries its own, specific complement of receptors and effectors. These phenotypical markers are quite often used to functionally and histochemically define a nociceptor . Note that sensory ganglia contain more than one type of nociceptor. Finally, there is the added complication of the temporal dimension of the neuronal responses to noxious stimuli: some are quite prompt, such as those triggered by burning of the nerve endings of the skin, while other influences work over extended periods of time, such as chronic underlying inflammation or axonal degeneration resulting from serious ongoing medical conditions such as diabetes. It seems obvious that more than one mechanism must be at work at the cellular and physiological levels for nociception to be accurate and reliable. It is also not surprising that being so complex and involving such a large number of molecular players, it can go wrong fairly often.
In the next sections we will briefly discuss some of the main aspects of the cellular biology and physiology of nociceptors and their link to pain. However, this is not intended as a full review and neither is it an in-depth description of the topics: it is only a guide to point out the most salient and active areas of interest and research in the field with some mechanistic insights and future perspectives.
The Nociceptors: Its Development and Maturation
Nociceptors develop from those neural crest stem cells that migrate from the dorsal part of the neural tube and form late during neurogenesis, whereas neurons born earlier become proprioceptors or low-threshold mechanoceptors [18–20]. All newly formed embryonic nociceptors express TrkA, the NGF receptor [20]. However, the transcription factors that determine nociceptor cell fate remain poorly understood. Differentiation of most TrkA+ neurons depends on the pro-neural transcription factor Neurogenin1 (Ngn1) [21, 22]. However, Ngn1 activity is not specific for nociceptors—it is also required for formation/differentiation of TrkB+ and TrkC+ cells, which eventually mature to become low-threshold mechanoceptors [21, 22]. However, the Runx1 runt domain transcription factor is expressed exclusively in TrkA+ neurons at early embryonic stages [23–26] but because its expression is initiated some time after the onset of TrkA expression, it is unlikely to be involved in early nociceptor cell fate determination [23]. Here it is important to mention that 1) some large, myelinated, fast conducting TrkA+ neurons become Aβ-nociceptors in adulthood, but are likely to have been TrkA- early in embryonic life; and 2) it is now clear that there is a degree of phenotypical switch in the dependence on trophic factors and hence its receptors (TrkA, TrkB, TrkC, etc.) happening between late embryonic and early postnatal stages [27–30]. It is also important to bear in mind that following sensory neurogenesis, potential nociceptors undergo two distinct differentiation pathways leading to the formation of two main classes of nociceptors: peptidergic and non-peptidergic nociceptors. These two sets of nociceptors express distinct repertoires of ion channels and receptors and innervate distinct peripheral and central targets [31–33]. This topic is briefly discussed in more detail in the next section, followed by a more in-depth description of the main differences existing between peptigergic and non-peptidergic nociceptors.
Segregation of Peptidergic Versus Non-peptidergic Nociceptors
During the perinatal and postnatal period, about one half of developing nociceptors switch off TrkA expression and start expressing Ret, the transmembrane signaling component of the receptor for glial cell-derived neurotrophic factor (GDNF) and other GDNF-related growth factors. The neurons undergoing this phenotypic switch in their trophic factor dependence become the so-called non-peptidergic nociceptors, most of which (>95 %) bind isolectin B4 (IB4+). The remaining nociceptors retain TrkA (a few also co-express Ret) and develop into the peptidergic class of nociceptors that do not bind IB4 and express the calcitonin gene-related peptides (CGRP) and substance P (SP) [34, 35]. The dynamic expression of Runx1 appears to be an important participant in this process [23, 24, 26]. Early embryonic nociceptors share a similar molecular identity, co-expressing both TrkA and Runx1 [23]. During the period when nociceptor segregation occurs, cells from Runx1 persist as nonpeptidergic neurons. Conditional knockout of Runx1 in the dorsal root ganglia (DRG) transforms these nonpeptidergic cells to a TrkA+ CGRP+ identity, and in this situation most nociceptors develop into peptidergic nociceptors [23, 26]. Conversely, constitutive expression of Runx1 in all nociceptors is sufficient to suppress embryonic peptidergic differentiation [24]. Runx1 also coordinates afferent targeting to the spinal cord; in mice that lack Runx1 prospective IB4+ non-peptidergic afferents adopt the projection pattern typical of peptidergic afferents [23]. These observations suggest that persistent Runx1 expression promotes the Ret+ nonpeptidergic cell fate, whereas loss of Runx1 is essential for peptidergic differentiation. Several studies have suggested that Runx1 and TrkA/Ret signaling pathways form a complex interaction loop for establishing nonpeptidergic nociceptor cell fate [36, 37]. TrkA-signaling is required to activate Ret, partly it appears by maintaining Runx1 expression at perinatal stages [36]. However, despite progress in teasing out the determinants of nociceptor specification, several issues remain to be resolved. Because both TrkA and Ret are required for afferents to innervate peripheral targets [36, 38], a loss of either TrkA or Ret signalling prevents nociceptors from receiving other target derived signals. Consequently, it is not known if TrkA signalling directly or indirectly controls expression of Runx1, and Ret, or if Ret signalling is directly involved in TrkA expression suppression. Additionally, while TrkA signalling is required to maintain Runx1 expression at embryonic stages, Runx1 expression is extinguished from TrkA+ peptidergic nociceptors during perinatal/postnatal development, therefore, we need to determine whether TrkA signaling switches from activating to suppressing Runx1 expression at different developmental stages or if a peripheral innervation defect in the absence of TrkA signalling indirectly extinguishes Runx1 expression. A further problem is that the intrinsic transcription factors that establish peptidergic nociceptor cell fate still remain elusive. Runx1 can, therefore, exert opposing activities depending on the cellular context. It will be extremely interesting to establish if changes in context-dependent transcriptional activities contribute to the phenotypic switches in nociceptors that occur in pathological conditions.
Subpopulations of Nociceptors
It is frequently stated that IB4 binding and TrkA expression define separate subpopulations of small, putative, nociceptive neurons. In the DRG, most small neurons (defined as those neurons showing slow action potential conduction velocity and a total cell area at the level of the nucleus of <400 μm2 in the rat—this cell area is different in other species). These small neurons express either TrkA or bind IB4 or both. It is therefore worth comparing these two populations. IB4 is a lectin from the plant Griffonia simplicifolia that binds to β-d-galactose residues in glycoconjugates on the cell surface and Golgi apparatus of small, neurofilament−poor DRG neurons in a variety of species [39]. That there is some degree of co-localization between TrkA expression and IB4 binding [40, 41] was confirmed with intracellular recording and dye injection studies in rat DRGs. These showed [1] that a third of C-fiber neurons were positive for both TrkA and IB4, with a tendency for reciprocal staining intensities for these two markers [2]. Most nociceptors strongly expressed TrkA or IB4 binding sites [3]. IB4 binding sites were present on C-fiber but not A-fiber nociceptive neurons, whereas TrkA expression was in both C- and A-fiber nociceptors [4]. Some weak positive labelling for TrkA and IB4 was seen in some D hair units. TrkA-, and not IB4-, positive neurons express SP and CGRP, as we stated in the previous section. Other differences between these neurons include projection of TrkA-positive neurons to laminae I and IIouter and IB4-positive neurons mainly to IIinner [42] of the dorsal horn [39] Compared with IB4-negative small neurons, IB4-positive neurons have longer duration action potentials and a smaller noxious heat-activated current [1], and the tetrodotoxin-resistant (TTXR) Na+ channel subunit Nav1.9 is preferentially expressed in IB4-positive cells [43]. It has also been shown that IB4-positive neurons selectively express the K+ leak channel TREK2, causing these neurons to be more hyperpolarized than IB4-negative nociceptors, and preventing these neurons from firing spontaneously [44], which is assumed to be the underlying cause for spontaneous pain [45, 46]. In summary, A-fiber nociceptors express TrkA but not IB4 binding sites, while most C-fiber nociceptors express one or the other, or both of these (Fig. 20.4). Apart from the NGF and GDNF dependence of TrkA expressing and IB4 binding neurons respectively, the functional differences between these groups of nociceptors are still relatively poorly understood.
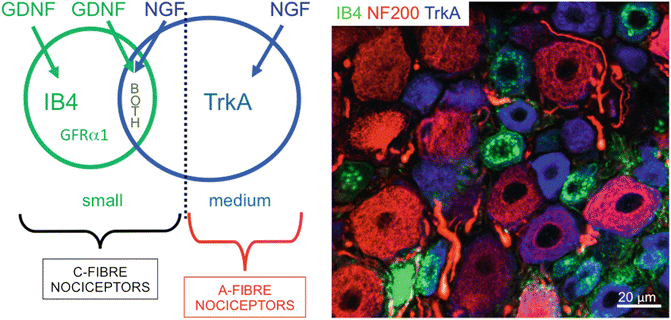
Fig. 20.4
Nociceptors are classified according to cell size (small, medium) and phenotype (binding of isolectin B4 or expression of the NGF receptor, TrkA) into C-fibre and A-fibre nociceptors. Note that IB4 binding small nociceptors express the GDNF-receptor GFR α 1. The right side panel shows triple immunofluorescence staining of a section of L5 normal rat DRG. Note the heterogeneity of subpopulations present in this small section of tissue. Neurons labelled with Neurofilament of 200 kDa (NF200, in red) are large and myelinated. Of these, a few express TrkA (in blue) which defines them as Aβ-nociceptors. Small neurons are either stained for TrkA or bind isolectin B4 (IB4, green) or rarely for both. All of these are nociceptors, unmyelinated and putative C-fiber neurons
Physiological Bases of Neuronal Excitability and Its Consequences for Pain
The mature nociceptor expresses dozens of ion channels and receptors, and the correct establishment of their expression is essential for nociceptors to detect specific noxious stimuli. There are two notable features about the developmental control of sensory channels/receptor expression. First, many sensory channels/receptors are expressed in only a partially overlapping or mutually exclusive manner, including TRP class thermal/chemical receptors and Mrg class G protein-coupled receptors [33, 47–49]. Second, the emergence of individual sensory channels/receptors is subject to complex temporal control. For example, expression of three TRP channels, TRPV1, TRPM8, and TRPA1, is initiated at E12.5, E16.5, and P0, respectively, while TRPA1 expression in peptidergic nociceptors is established at P0 and non-peptidergic nociceptors at P14, respectively [49]. Albeit of considerable interest in basic research, these complex developmental processes are beyond the scope of this chapter and have already been thoroughly reviewed in the literature [17].
As stated above, the mature nociceptor expresses a large number of ion channels and other associated receptors. These ion channels are carried through the cell membrane ion currents that are responsible for the excitability of the neuron. They play a pivotal role as direct or indirect controllers of or contributors to action potential generation and propagation along the nerve fibers. It is therefore important for us to look at the growing body of knowledge accumulated about these ion channels and to discuss the implications that their electrophysiological properties have for pain physiology normally and in models of chronic pain.
Na+ Currents and Channels
Voltage-gated sodium channels (called Nav) are essential for generation and conduction of action potentials. The currents are subdivided into tetrodotoxin-sensitive (TTXS) and TTXR. The channels designated Nav1.1, 1.2, 1.3, 1.6, and 1.7 are TTXS, while Nav1.5, 1.8, and 1.9 are TTXR. Three of these proteins, Nav1.8 and Nav1.9 (both TTXR), and Nav1.7 (TTXS) are preferentially (but not exclusively) expressed in small DRG neurons. Described next were the main known properties of these channels and how their function and/or expression are altered in models of inflammation and nerve injury.
Nav1.8 (Also Called SNS/PN3)
Nav1.8 is expressed in rats in small- to medium-sized DRG neurons [50] most of which are nociceptive [51]. Nav1.8 gives rise to a TTXR Na+ current that is believed to contribute the majority of the inward Na+ current in action potentials of small DRG neurons and it is most likely responsible for the TTXR current at nociceptive receptor terminals [52, 53]. Its electrophysiological properties probably contribute to properties of nociceptive neurons, including its high activation threshold of about −35 mV. Consider that normal resting potential in C-fiber nociceptors is ~55 mV and therefore a threshold of −35 mV implies that a neuron must be made 20 mV more positive in order to fire an action potential. This is a significant change in membrane potential and it suggests that such a nociceptor will not fire easily unless the intensity of the stimuli is high enough to be registered as a threat or it is noxious. The slow kinetics of Nav1.8 give rise to long-duration action potentials, and contributes to the large action potential overshoot, and its rapid repriming. All this may enable firing even in depolarized fibers [50–52, 54]. In essence, the slow inactivation of Nav1.8 means that these channels may be able to sustain repetitive firing even at depolarized membrane potentials. That is why they are thought to underlie spontaneous pain which requires ongoing firing in C-fiber neurons at very slow rates [45].
Inflammation and Nerve Injury
Several inflammatory mediators can acutely (minutes to hours) decrease the activation threshold, and/or increase the kinetics or magnitude of the TTXR Na+ current (presumed to be Nav1.8 related) [55] and may contribute to acute hypersensitivity at nerve terminals. In the longer term, Nav1.8 mRNA and TTXR Na+ current in small DRG neurons and in cutaneous fibers are upregulated during most studies on inflammation [56, 57]. In some models of neuropathic pain, such as 7-day axotomy of the L5 nerve, TTXR current density and Nav1.8 mRNA and protein are decreased [58]. This reduction in Nav1.8 may explain why axotomized C-fiber neurons are incapable of action potential firing despite being relatively depolarized [44].
Nav1.9
Nav1.9 is also known as NaN/SNS2. Similarly to Nav1.8, Nav1.9 gives rise to a TTXR current. Nav1.9 immunoreactivity is mostly present Sin small DRG neurons [59, 60], preferentially in those with IB4 binding [43] (and therefore, non-peptidergic) and along C-fibers and at nodes of Ranvier of thinly myelinated fibers [61]. It gives rise to a persistent, depolarizing TTXR Na+ current in small DRG neurons as a result of its low activation threshold and ultra-slow inactivation [62], thus it is likely to influence membrane excitability, although the magnitude and mode of this effects remain poorly understood. Interestingly, GDNF, but not NGF, upregulates Nav1.9 mRNA [43]. This is in agreement with GDNF being the main trophic factor required to maintain the phenotype of IB4-binding small C-nociceptors in vivo [41]. Interestingly, Nav1.9 expression is linearly and positively correlated with TREK2 expression (a K+ leak channel, see below) in DRG neurons, which suggests that both channels are part of the same control mechanism of neuronal excitability in IB4-binding neurons [44].
Inflammation and Nerve Injury
Nav1.9 mRNA in DRG neurons is increased after inflammation [59], after exogenous GDNF administration [43], and it is also activated by the application of a cocktail of pro-inflammatory mediators [63]. Axotomy induces a decrease in Nav1.9 mRNA and protein in the DRG [43, 58, 59] that is reversed in IB4-positive neurons by exogenous GDNF [43]. A lack of function mutation of the gene encoding for Nav1.9 in humans (SCN11A) has recently been reported [12, 14]. Patients with this rare mutation experience lack of pain, and are prone to suffer extensive burns. This highlights the importance of the protective role of acute pain which prevents us from suffering life-threatening injuries.
Nav1.7 (or PN1)
Nav1.7 is expressed more highly in small rather than large DRG neurons [64] despite the fact that its mRNA is present in neurons of all sizes [65]. It is thought to carry much of the TTXS inward current in action potentials in small neurons [66]. Nav1.7 protein is present in fibers and terminals of cultured DRG neurons [67]. Its slow inactivation, combined with its low activation threshold (closer to −50 mV) [66], may be important in the generation of receptor potentials and contributing to the generation of action potentials. NGF causes a long-lasting (weeks) increase in Nav1.7 protein in DRG neurons in vivo [68].
Nav1.7 expression drops substantially after axotomy while essentially remaining unchanged after acute cutaneous inflammation [44]. The role of this channel in pain is normally emphasized by the finding that a lack-of-function mutation of its gene derives a lack of pain sensitivity and the consequent exposure to injury (e.g., breaking bones). This seems to be a genetic trait that is inherited [10, 69, 70]. It has also been shown that a scorpion toxin causes a gain of function of Nav1.7 leading to pain hypersensitivity [71].
It is important to note that certain types of pain (e.g., oxaliplatin-induced neuropathy), do not require either Nav1.7 or Nav1.8 [72]. Thus, proper patient stratification and accurate diagnosis is essential to correctly treat chronic pain using modulators of Na+ channel function.
Other Na+ Subunits
mRNAs of several other TTXS subunits are more abundant in medium and large neurons. These include Nav1.1, 1.2, and 1.6 and Nav2.2 (NaG) [73]. Following axotomy, the appearance of a more rapidly repriming TTXS current is thought to contribute to hyperexcitability in axotomized small DRG neurons in vitro [74, 75]. This has been ascribed to increased expression of Nav1.3 (also known as brain type III), but roles for other TTXS channels have not been ruled out.
Na+ Channel β Subunits
Na+ channel β subunits may interact with the cytoskeleton or extracellular matrix and play roles in Na+ channel trafficking within cells and insertion into membrane, thought to be mediated by annexins and the auxiliary protein p11 [76–78]. When co-expressed with β subunits, subunits can alter the kinetics, peak current, and/or voltage dependencies of β subunits [79].
K+ Currents and Channels
K+ channels are central to the control of resting membrane potential, after-hyperpolarization, and firing frequency and they influence adaptation. They tend to increase membrane potential stability (i.e., less likely to oscillate), and at least some of the K+ channels that contribute to long duration after-hyperpolarizations may prove to be more highly expressed in nociceptors. Despite much work in this field in recent years, there is still a lack of complete understanding of two main questions. 1) Which K+ channels are expressed by different functional subpopulations of primary afferent neurons, and 2) How these channels work together to maintain membrane potential stability and to provide re-polarization/after-hyperpolarization in firing nociceptors [80, 81].
Voltage-Gated K+ Currents and Kv Channels
The two main groups of calcium-insensitive voltage-gated K+ currents are the depolarization-activated delayed rectifier (IKv) and fast transient (IA) currents. The protein subunits of the channels that underlie these currents are the Kv subunits.
Delayed Rectifier Currents
Fast Transient (A-Type) K+ Currents
Fast transient K (IA) currents tend to clamp resting potential at hyperpolarized voltages until they inactivate, thus prolonging the after-hyperpolarization and slowing/preventing repetitive discharges [85], IA currents are present in both large cutaneous afferents and small DRG neurons [84] but are more prominent in slowly conducting afferents [86]. IA can be subdivided into rapidly (fast IA) and slowly [87] inactivating types [88]. Slow IA is particularly prominent in small DRG neurons with TTXR action potentials (see [88]), and may therefore contribute to the broad after-hyperpolarizations of nociceptive neurons.
In DRG neurons IKv and slow IA are both sensitive to dendrotoxin, but fast IA is not [88]. Kv1.1 and 1.2 are associated with the delayed rectifier, whereas Kv1.4 is associated with fast IA [89, 90], and it has been suggested that Kv1.1/1.2 associated with Kv1.4 (dendrotoxin insensitive) may give rise to the slow IA in DRG neurons [88, 89], Kv1.1, 1.2, 1.3, 1.4, 1.5, and 1.6 and Kv_2.1 are all expressed in DRG neurons; of these, Kv1.1 and 1.2 are more abundant and highly expressed in medium- to large-sized neurons, and Kv_2.1 is also more highly expressed in medium to large neurons, whereas Kv1.4 is more highly expressed in small neurons [88, 89, 91]. Kv1.1 has recently been shown to play a central role in mechanosensation [92].
M Currents
M currents (IM) are voltage and time dependent, noninactivating, and activated at negative voltages (beginning at approximately −70 mV). Their inhibition by acetylcholine or other agents leads to increased neuronal excitability [93]. IM are associated with KCNQ2/3 and −5 subunits [93–95], all members of the six transmembrane superfamily that are distantly related to Kv channels. IM as well as KCNQ2, −3, and −5 have been detected in both small and large DRG neurons [94, 96]. Blocking of IM with linopirdine causes increased firing in response to current injection in small DRG neurons, indicating that the IM may normally act as a brake to firing in these neurons. After cutaneous inflammation, there is inhibition of IM leading to depolarization and exacerbated firing [97]. More recently, IM present in nociceptors (probably including peptidergic and non-peptidergic C-neurons) have been attributed to the expression of Kv7.2 [97, 98] and Kv7.5 [8, 99], although evidence is contradictory and requires further study.
Inwardly Rectifying K+ Channels
Inwardly rectifying K+ (Kir) channel [100] current contributes to the resting membrane potential in a number of cell types, including DRG neurons, where Kir current is mostly in medium-size neurons [101]. Little is known about Kir-related channel subunits in DRGs. However, there is evidence that paclitaxel reduces the expression of Kir1.1 and Kir3.2 in sensory neurons, leading to neuropathic pain and nociceptor hiperexcitability [102]. Kir3.2 has also been involved in the response to opioids [103]. Other Kir channels (2.1, 2.2 and 2.3) have been proposed as key controllers of the pacemaker activity of lamina I spinal cord neurons part of the pain circuit [104].
Ca2+-Activated K+ Currents
Ca2+-activated K+ currents (IKCa) are of three types, related to BK [87], IK, and SK channels (big, intermediate, and small conductance channels, respectively), all activated by elevated intracellular Ca2+; BK is also voltage dependent. Functionally BK is associated with after-hyperpolarizations that develop rapidly and decay in 10–100 ms, and SK with slower after-hyperpolarizations that may last for seconds and limit firing frequency [105, 106]. Either or both of these could therefore contribute to the long after-hyperpolarization in nociceptors, although this remains to be established. BK and SK currents are found in DRG neurons [107–109], with BK currents observed in two thirds of small DRG neurons. Immunoreactivity for SK1 and IK1 channels was found in many human and rat DRG neurons [110]. A related channel, SLACK (sequence like a Ca2+-activated K+), is expressed in rat DRG neurons [111]. SLACK has a conductance similar to that of BK, with which it can interact to generate an I-KCa channel (different from IK1). Interestingly, SLACK (and its partner, SLICK) is required for the depolarization of afterpotential in medium DRG neurons [112]. Protein kinase A induced internatilization of SLACK causes neuronal hyperexcitability [113]. It has been demonstrated that a reduced SLACK expression leads to increased thermal and mechanical sensitivity, a process regulated by the chloride channel TMEM16C [114].
Related posts:
 Knowledge, by Philosophy or Science? Psychotherapy or Neuroscience?
Neurovascular Cognitive Alterations: Implication of Brain Renin–Angiotensin System
Animal Models of Psychopathology and Its Relation to Clinical Practice
Adenosine in the Neurobiology of Schizophrenia: Potential Adenosine Receptor-Based Pharmacotherapy
Mindfulness and Neuroimaging
Biological Markers in Psychiatry and Its Relation with Translational Approaches: Brief Historical Review
Knowledge, by Philosophy or Science? Psychotherapy or Neuroscience?
Neurovascular Cognitive Alterations: Implication of Brain Renin–Angiotensin System
Animal Models of Psychopathology and Its Relation to Clinical Practice
Adenosine in the Neurobiology of Schizophrenia: Potential Adenosine Receptor-Based Pharmacotherapy
Mindfulness and Neuroimaging
Biological Markers in Psychiatry and Its Relation with Translational Approaches: Brief Historical Review
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


