28
CHAPTER
![]()
Third-Generation Antiepileptic Drugs
Cesar C. Santos
The U.S. Food and Drug Administration (FDA) approved the third-generation antiepileptic drugs (AEDs) between 2004 and 2012. The AEDs included in this category include pregabalin, lacosamide, rufinamide, vigabatrin, clobazam, ezogabine, and perampanel. Though many of these are truly new compounds, some like vigabatrin and clobazam have been available outside the United States long before they were approved for use in the United States. This chapter focuses on these AEDs, and a summary of their various properties is presented in Table 28.1 (located at the end of the chapter).
PREGABALIN
Indications
Pregabalin is an AED used as an adjunct therapy for partial seizures with or without secondary generalization and in the management of patients with neuropathic pain associated with diabetic peripheral neuropathy and spinal cord injury, postherpetic neuralgia, and fibromyalgia. In 2007, pregabalin became the first medication that was FDA approved for the treatment of fibromyalgia. Although not FDA approved for this indication, it has been found effective in treating patients with generalized anxiety disorder.
Dosing
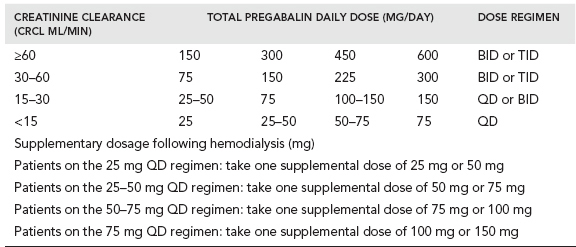
The available preparations of pregabalin include capsules of 25, 50, 75, 100, 200, 225, and 300 mg and an oral solution containing 20 mg/ml. Pregablin is given orally with or without food. If it needs to be discontinued, gradual tapering over a minimum of 1 week is recommended. The usual starting dose is 150 mg/day (either 50 mg three times a day or 75 mg twice a day). Since pregabalin is primarily eliminated by renal excretion, dose adjustment is recommended based on renal function (Table 28.2).
A dose of 150 mg to 600 mg per day of pregabalin has been shown to be effective as adjunct treatment of partial seizures with or without secondary generalization in adults. Efficacy and adverse events are both dose related. The recommended starting dose is 150 mg/day (either 75 mg given twice a day or 50 mg given three times a day). Based on response and tolerability, the dose can be adjusted to a maximum dose of 600 mg/day.
When used for neuropathic pain associated with diabetic peripheral neuropathy, the recommended maximum dose of pregabalin is 100 mg given three times a day in patients with creatinine clearance of at least 60 ml/min. For neuropathic pain associated with spinal cord injury, the recommended dose is 150 mg to 600 mg per day. In postherpetic neuralgia the recommended dose is 75 mg to 150 mg given twice a day or 50 mg to 100 mg given three times a day (150 mg to 300 mg/day) in patients with creatinine clearance of at least 60 ml/min. The recommended dose for fibromyalgia is 300 mg to 450 mg per day. A dose above 450 mg/day is not recommended.
Pharmacology
In the central nervous system, pregabalin binds to the alpha2-delta subunit of the voltage-dependent calcium channel. It decreases the release of several neurotransmitters, including glutamate, norepinephrine, substance P, and calcitonin gene-related peptide, which may account for its ability to reduce neuronal excitability and seizures in vivo.
Peak plasma concentration of pregabalin occurs within an hour when taken on an empty stomach. When taken with food, pregabalin decreases Cmax by 25% to 30% and Tmax by 2.5 hours. However, food does not affect the extent of absorption. Pregabalin is not bound to plasma proteins. The volume of distribution is 0.56 l/kg. Half-life is 6.3 hours. Metabolism of pregabalin in humans is negligible. Major metabolite is N-methyl pregabalin. About 98% of pregabalin is excreted unchanged in the urine. Renal clearance is 73 ml/minute.
Efficacy Data
Three randomized, double-blind, placebo-controlled studies involving a total of 1,052 patients with refractory partial seizures, 12 years and older, showed significant improvement in seizure control at 150, 300, and 600 mg/day with a clear dose–response relationship. Almost 50% of patients receiving 600 mg/day achieved greater than or equal to 50% seizure reduction. There was no difference between a twice-a-day (BID) and three times-a-day (TID)dosing schedule. Efficacy became evident as early as the first week of treatment (1).
Efficacy in Other Conditions
In patients with neuropathic pain associated with diabetic peripheral neuropathy, a 6-week, double-blind, placebo-controlled trial involving 246 patients showed significant improvement in mean pain score (4.3 vs. 5.6 for placebo, P = .0002) and an increase in the number of patients with greater than or equal to 50% decrease from baseline pain (39% vs. 15% for placebo, P = .002) at 600 mg/day dose schedule (2).
A 12-week, multicenter, placebo-controlled trial was conducted in patients with neuropathic pain associated with spinal cord injury. A total of 137 patients were randomized to either placebo or flexible dose of pregabalin (150 to 600 mg/day) while continuing on their stable pain medication. The primary endpoint was the endpoint mean pain score (last 7 days daily pain diary entry). The endpoint mean pain score for the pregabalin-treated group was 4.62 compared with 6.27 for the placebo group (P <.001). Treatment response was seen as early as week one and maintained throughout the stabilization phase. Pregabalin treatment was also associated with significant improvement in sleep (3).
In a postherpetic neuralgia trial, pregabalin-treated patients receiving either 600 mg/day (CrCl >60 ml/min) or 300 mg/day (CrCl 30–60 ml/min) showed significant improvement in endpoint mean pain scores (mean of the last seven daily pain ratings), 3.60 vs. 5.29 for placebo (P = .0001). In addition, patients receiving pregabalin had significant improvement in sleep (P = .001) (4).
Fibromyalgia treatment trials also showed improvement with pregabalin. Placebo was compared with 150, 300, and 450 mg/day dose of pregabalin in an 8-week trial looking at pain, sleep, fatigue, and health-related quality of life (QOL) in 529 patients with fibromyalgia syndrome. The 450 mg/day of pregabalin arm resulted in (a) significant improvement in pain severity, (b) significantly more patients with greater than or equal to 50% improvement in pain at the end point, and (c) significant improvement in sleep quality, fatigue, and several domains of health-related QOL (5).
Adverse Effects
The most common side effects of pregabalin are dizziness, drowsiness, dry mouth, edema, blurred vision, weight gain, and difficulty concentrating. Other side effects include thrombocytopenia and increased creatinine kinase. Rarely, pregabalin has been associated with angioedema.
Toxicity, Overdose, and Contraindications
Patients with severe renal failure on pregabalin may develop myoclonus as a result of gradual drug accumulation.
Warnings and Precautions
As with all other AEDs, suicidal behavior and ideation are listed as possibly occurring with pregabalin. Patients should be monitored for signs and symptoms of depression or worsening depression, suicidal ideation or behavior, and/or unusual changes in mood or behavior.
Teratogenicity
There are no adequate studies of pregabalin in pregnant women. Pregabalin is a Pregnancy Category C drug. Animal studies have shown increased incidence of fetal structural abnormalities as well as other manifestation of developmental toxicity like lethality, growth retardation, and functional impairment of both nervous and reproductive systems.
Pregabalin is excreted in breast milk of rats. Although it is unknown whether this is true in humans, pregabalin use is not recommended while breastfeeding.
Special Safety Concern
Drugs that depress the central nervous system (CND), including alcohol, may increase the sedative effects of pregabalin. Pregabalin can cause rhabdomyolysis, and, as noted earlier, rarely can cause angioedema.
Drug Interactions
Owing to the pharmacokinetics of pregabalin, it is unlikely to be affected by other drugs through metabolic interactions or protein binding displacement. There are no reported pharmacologic interactions in vivo. However, it may have some potential interaction with opiods, benzodiazepines, barbiturates, ethanol, and other CNS depressant medications. It is a Schedule V drug, classified as a CNS depressant. Use of pregabalin with pioglitazone (Actos) and rosiglitazone (Avandia) may cause weight gain, fluid retention, and possibly heart failure.
Use in Special Population
Since pregabalin is eliminated primarily by renal excretion, the dose needs to be adjusted in patients with reduced renal function. Dose adjustment is calculated based on creatinine clearance (Table 28.2). The use of pregabalin in preclinical trials in patients with diabetic neuropathy and epilepsy failed to show liver toxicity. However, postmarketing use has shown a rare instance of mild liver injury without jaundice with onset of symptoms within 3 to 14 days. Both cholestatic and hepatocellular patterns have been reported. The mechanism of injury is presumed to be idiosyncratic. The safety and efficacy profile of pregabalin is similar in the elderly and younger patients.
Pediatric Use
The safety and efficacy of pregabalin in pediatric patients have not been established.
LACOSAMIDE
Indications
Lacosamide is indicated as an adjunct treatment of partial seizures in patients 17 years of age and older. Recently, it was also approved for use in monotherapy in patients with partial seizures aged 17 years and older.
Dosing
Lacosamide is available as tablets of 50 mg, 100 mg, 150 mg, and 200 mg concentrations, injections of 10 mg/ml for intravenous infusion and oral solution of 10 mg/ml. It may be taken with or without food. For treatment of partial-onset seizures, lacosamide can be started via either an oral or intravenous administration. The initial recommended dose is 50 mg twice a day. The dose can be increased weekly by 100 mg up to the recommended maintenance dose of 200–400 mg/day based on response and tolerability.
When lacosamide needs to be switched from oral to IV dosing, the initial IV dose should be equivalent to the total daily dose and frequency of oral dosing. Each IV dose should be infused over a period of 30 to 60 minutes. When switching IV lacosamide to oral dosing, the oral dose should be equivalent to the total daily IV dose and administered twice daily.
Pharmacology
The exact mechanism of action of lacosamide is unknown. In vitro studies have shown that it selectively enhances slow inactivation of voltage-gated sodium channels, which results in stabilization of hyperexcitable neuronal membranes and inhibition of neuronal firing. Previously it was also thought to bind to collapsing response mediator protein-2 (CRMP-2), a phosphoprotein mainly expressed in the nervous system and known to be involved in neuronal differentiation and control of axonal outgrowth. Recent data suggest that at therapeutic doses this may not occur.
Lacosamide is completely absorbed. Both the rate and extent of absorption are not affected by food. Bioavailability of orally administered lacosamide is about 100%. The elimination half-life is 13 hours and steady state is reached after 3 days of twice-a-day dosing. The peak plasma concentration is reached at the end of IV infusion and in 1 to 5 hours following oral administration. The volume of distribution is about 0.6 L/kg. Protein binding is less than 15%, and there is a linear relationship at a dose range of 100 to 800 mg/day. Elimination is primarily by renal excretion with 40% of the drug being excreted in the unchanged form. Metabolism of lacosamide is via the CYP2C19 system. Although peak concentrations are unchanged, in patients with mild–moderate renal impairment (creatinine clearance [CrCl] of 30–80 ml/min) and in patients with severe renal impairment (CrCl of 30 ml/min or less), the area under the curve (AUC) for lacosamide is increased by 25% and 60%, respectively. Dose adjustments are recommended. Dose adjustment is also recommended following hemodialysis because there can be as much as 50% reduction in the lacosamide AUC. Compared with healthy volunteers, the AUC of lacosamide is 50% to 60% higher in patients with moderate hepatic impairment. Dosage adjustment is also recommended in patients with mild–moderate hepatic impairment. Use of lacosamide in patients with severe hepatic impairment is not recommended.
Efficacy Data
Efficacy data for lacosamide as adjunctive therapy for partial-onset seizures comes from three randomized, double-blind, placebo-controlled, multicenter studies. All studies involve adult patients with medically refractory partial seizures on one to three AEDs. Study designs were similar including the inclusion criteria and dose of lacosamide. A 50% reduction in seizure frequency was comparable among the three studies, 38% to 41%, at 400 mg and 600 mg/day dose given twice a day, which was statistically superior compared to placebo (6–8). In addition, an open-label extension study showed a 50% or greater reduction in seizures in 46.6% of patients. A conversion to monotherapy trial demonstrated that patients treated with lacosamide 300 mg and 400 mg/day dose exited the trial less often than historical controls (9).
Efficacy in Other Conditions
Lacosamide has been studied in painful diabetic neuropathy, but is not currently approved for used in that indication.
Adverse Effects
The most common adverse effects of lacosamide are diplopia, headache, dizziness, and nausea.
Toxicity, Overdose, and Contraindications
There are no absolute contraindications noted for lacosamide.
Warning and Precaution
As with all AEDs, suicidal behavior and ideation can occur with lacosamide. It can cause dizziness and ataxia. The utility of lacosamide should be seriously evaluated in patients with cardiac conduction problems like second-degree atrioventricular (AV) block, those who are taking drugs known to cause prolongation of the PR interval, and patients with severe cardiac disease. Lacosamide may also cause syncope. It should be withdrawn gradually in patients with epilepsy to minimize the potential of increased seizure frequency. Rare cases of multi-organ hypersensitivity reactions have been reported.
Teratogenicity
There is no adequate data from the use of lacosamide in pregnant women. It should be used during pregnancy when its benefits clearly outweigh the unknown risks. There are also no data whether lacosamide is excreted in human breast milk. It is recommended that breastfeeding be discontinued during use of lacosamide.
Special Safety Concern
Although peak concentrations are unchanged, in patients with mild–moderate renal impairment (CrCl of 30–80 ml/min) and in patients with severe renal impairment (CrCl of 30 ml/min or less), the area under the curve (AUC) for lacosamide is increased by 25% and 60%, respectively. Dose adjustments are recommended. Dose adjustment is also recommended following hemodialysis because there can be as much as 50% reduction in the lacosamide AUC. Compared with healthy volunteers, the AUC of lacosamide is 50% to 60% higher in patients with moderate hepatic impairment. Dosage adjustment is also recommended even in patients with mild–moderate hepatic impairment. Use in patients with severe hepatic impairment is not recommended.
Drug Interaction
There is no known relevant drug–drug interaction between lacosamide and common AEDs.
Use in Special Population
The use of lacosamide in patients with renal and hepatic impairment has been discussed earlier. In geriatric use, the AUC was 30% and 50% higher in men and in women, respectively, over the age of 75 years.
Pediatric Use
Lacosamide is not approved for use in children under age 17 years.
RUFINAMIDE
Indication
Rufinamide is indicated as an adjunct treatment of drop attacks associated with Lennox-Gastaut syndrome (LGS).
Dosing
Rufinamide is available for oral administration in 200 mg and 400 mg scored, film-coated tablets. It is also available in a liquid (40 mg/ml) formulation. In children 4 years and older with LGS, rufinamide should be started at a daily dose of 10 mg/kg/day given in two equally divided doses. The dose can be increased by 10 mg/kg every other day to a target dose of 45 mg/kg/day or 3,200 mg/day, whichever is less, administered in two equally divided doses. In adults with LGS, rufinamide should be started at a daily dose of 400–800 mg/day given in two equally divided doses. It can be increased by 400–800 mg every other day to a maximum dose of 3,200 mg/day, administered in two equally divided doses. Rufinamide should be given with food.
Pharmacology
Rufinamide is a triazole derivative. The exact mechanism of action is unknown. In experimental models, it has been shown to suppress hyperexcitability of neurons by prolonging the inactivation phase of the voltage-gated sodium channels. At relatively high concentrations, rufinamide has an inhibitory effect on mGluR5 receptor subtype.
The peak plasma concentration (Tmax) for rufinamide is reached between 4 and 6 hours with or without food. The rate of absorption and degree of exposure, however, can be maximized if rufinamide is taken with food. Plasma proteins do not extensively bind it. However, the absorption rate is very slow with peak plasma concentration occurring 4 to 6 hours after oral intake. It is estimated that greater than 85% of an orally administered 600 mg dose is absorbed in healthy volunteers. Absorption decreases with increasing dose.
The elimination half-life is between 6 and 10 hours. Steady state is reached within 2 days. Rufinamide pharmacokinetics is not affected by impaired renal function. However, patients on dialysis may experience up to 30% reduction in exposure so dose adjustment may have to be considered. Only about 34% of rufinamide is bound to plasma proteins, and so the potential for drug–drug interaction through displacement is very small. The volume of distribution is dose dependent.
Rufinamide is metabolized via hydrolysis by carboxylesterases to a pharmacologically inactive metabolite, carboxylic acid derivative, which is excreted in the urine. It is not metabolized by the cytochrome P450 but is considered a weak inhibitor of CYP2E1 and a weak inducer of CYP3A4. Elimination half-life is between 6 and 10 hours. Rufinamide is primarily excreted via the kidneys.
Efficacy Data
Rufinamide was tested in a randomized, double-blind, placebo-controlled study, which showed a median reduction in the frequency of total seizures (32.7% vs. 11.7%, P = .0015) and tonic–atonic seizures (42.5% reduction vs, 1.4% increase, P < .0001) in patients receiving drug compared with patients on placebo (10). A large Phase 2, double-blind, randomized, placebo-controlled study showed a significant, linear dose–response relationship (P = .003) at doses of 200, 400, 800, and 1,600 mg/day (11). Rufinamide was also tested in adults with partial-onset seizures, but the drug does not have an indication for partial-onset seizures in adults.
Efficacy in Other Conditions
Rufinamide has not been tested in detail in conditions other than epilepsy.
Adverse Effects
The most commonly reported adverse reactions that occur with rufinamide include headaches, dizziness, fatigue, somnolence, and nausea
Toxicity, Overdose, and Contraindications
Rufinamide is contraindicated in patients with familial short QT syndrome.
Warning and Precaution
As with all other AEDs, rufinamide can cause suicidal behavior and ideation. It has been associated with multiorgan hypersensitivity reactions. Caution should be exercised when using rufinamide concomitantly with other drugs that shorten the QT interval. Rufinamide should be withdrawn slowly to minimize the risk of withdrawal seizures, seizure exacerbation, or status epilepticus.
Teratogenicity
Rufinamide is a Pregnancy Category C AED. Studies in animals reveal no teratogenic effects. There are no clinical data on exposed pregnancies. However, rufinamide should not be taken during pregnancy and in women of childbearing age not using contraceptive measures unless clearly necessary. Although there are no data to suggest that rufinamide is excreted in breast milk, breastfeeding should be avoided during maternal use of rufinamide due to potential harmful effects.
Special Safety Concerns
A relatively unique safety issue with rufinamide is that it has been shown to shorten the QT interval on electrocardiography (ECG). For this reason, it is contraindicated in patients with familial short QT syndrome. In addition, it should be used with caution in patients taking other medications with the same effect. As noted earlier, rufinamide can cause multiorgan hypersensitivity reactions like drug reactions with eosinophilia and systemic symptoms (DRESS) and Stevens-Johnson syndrome.
Drug Interaction
Rufinamide has been shown to increase the serum phenytoin by as much as 21% with potential for further increases due to the dose-dependent pharmacokinetics of phenytoin. It also increases the clearance of carbamazepine and lamotrigine and decreases the clearance of phenobarbital. Valproic acid can increase the serum concentration of rufinamide by as much as 70% in children. It is recommended to decrease the dose of rufinamide to less than 400 mg in adults and by 50% to 60% in children when used concomitantly with valproic acid. It should be titrated upward slowly when starting rufinamide as an add-on AED to valproic acid. Rufinamide can decrease the effectiveness of oral contraceptives with ethinyl estradiol and norethindrone.
Use in Special Population and Pediatric Use
The pharmacokinetics of rufinamide is similar in patients with severe (CrCl < 30 ml/min) renal impairment and that of healthy volunteers. However, dose adjustment is recommended when using rufinamide during dialysis. Although there are no specific studies addressing the effect of rufinamide in patients with hepatic impairment, its use is not recommended in patients with severe hepatic impairment. Rufinamide clinical studies did not include sufficient number of patients 65 years and older to determine if there are particular efficacy and safety considerations in this population.
Pediatric Use
Rufinamide is indicated as an adjunctive treatment in children with seizures secondary to LGS. Its use in children less than 4 years has not been established. In children over age 4 years, the pharmacokinetics of rufinamide is similar to that in adults.
VIGABATRIN
Indication
Vigabatrin is indicated for the treatment of infantile spasms (IS)(in children aged 1 month–2 years of age) and as adjunctive treatment of refractory complex partial seizures in adults for whom the benefits outweigh the potential risk of vision loss.
Dosing
Vigabatrin is available in powder form for oral solution (500 mg) and in tablet form (500 mg). It should be given in two divided doses starting at an initial dose of 50 mg/kg/day. This can be titrated slowly (25–50 mg/kg/day) up to a maximum dose of 150 mg/kg/day.
Pharmacology
The exact mechanism of action of vigabatrin is unknown. It is presumed to exert its anticonvulsant property by irreversibly inhibiting gamma-aminobutyric acid transaminase (GABA-T) resulting in increased levels of gamma-aminobutyric acid (GABA), the main inhibitory neurotransmitter in the CNS.
Vigabatrin has a linear pharmacokinetic pattern with a half-life of about 7.5 hours. Following oral administration, it is absorbed completely with a Tmax of 1 hour following a single as well as multiple doses. When taken with food, the Cmax is decreased by 33%, Tmax is increased to 2 hours, and the AUC is unchanged. Vigabatrin does not bind to plasma proteins. The volume distribution for vigabatrin is 1.1 L/kg. It induces CYP2C9. Vigabatrin is not significantly metabolized and is primarily eliminated via the kidneys.
Efficacy Data
Vigabatrin’s efficacy data for the treatment of medically refractory complex partial seizures was derived from two separate randomized, double-blind, placebo-controlled studies. One study compared the efficacy and safety of vigabatrin up to 3 g/day dose with placebo in a double-blind, placebo-controlled fashion. A total of 182 patients were enrolled. This study showed that 3 g of vigabatrin was not only well tolerated, but it was significantly more effective than placebo as an add-on therapy for complex partial seizures (12). The second study was a dose–response study comparing 1 g, 3 g, or 6 g of vigabatrin to placebo. This study showed significant improvement in seizure control compared with placebo (≥50% reduction in seizure frequency of 7% for placebo vs. 24%, 51%, and 54% for 1 g, 3 g, and 6 g doses, respectively). The 6 g/day dose was not significantly different compared with 3 g/day dose (13).
Two studies were used to evaluate vigabatrin’s effectiveness in IS for FDA approval (14). The first study was a multicenter, randomized study comparing low-dose (18–36 mg/kg/day) and high-dose (100–148 mg/kg/day) vigabatrin in patients with both symptomatic and cryptogenic infantile spasms. Seventeen (16%) infants treated with high-dose vigabatrin became spasm free compared with only 7% in the low-dose group. Another study involved 40 patients. It was a multicenter, randomized, double-blind, placebo-controlled study in which infants were started on 50 mg/kg/day and based on response, titrated to 150 mg/kg/day. The end point was the percent change in spasm frequency. There was no difference in primary end point. However, a posthoc review showed a significant overall reduction in spasms in the treatment group (68.9% vs. 17% in the controls).
Efficacy in Other Conditions
Vigabatrin has not been tested in detail in conditions other than epilepsy.
Adverse Effects
The most commonly reported adverse effects include headache, somnolence, fatigue, dizziness, convulsion, nasopharyngitis, weight gain, upper respiratory infection, visual field defect, depression, tremor, nystagmus, nausea, diarrhea, memory impairment, insomnia, irritability, abnormal coordination, blurred vision, diplopia, vomiting, influenza, pyrexia, and rash.
Toxicity, Overdose, and Contraindications
Vigabatrin overdose has not resulted in death. However, it has caused coma, unconsciousness, and/or drowsiness. There is absolute contraindication to the use of vigabatrin.
Warnings and Precautions
Vigabatrin can result in suicidal ideation and behavior, as can other AEDs. It should be withdrawn slowly to minimize the risk of withdrawal seizures, seizure exacerbation, or status epilepticus. Vigabatrin has been shown to cause peripheral neuropathy in adults. It can also cause weight gain and peripheral edema. Vigabatrin can cause visual changes, abnormal MRI findings, and neurotoxicity. These are discussed further.
Teratogenicity
No significant data regarding the effects of vigabatrin on pregnancy are available. Vigabatrin is excreted in breast milk.
Special Safety Concerns
Vigabatrin can cause permanent vision loss in infants, children, and adults. The extent of vision loss in infants and children is not well characterized. In adults, it causes concentric constriction of the visual filed in over 30% of cases. In some cases, it can cause central retinal injury. The onset is unpredictable. It can occur within weeks of the start of treatment or at any time thereafter. Visual loss may worsen even after vigabatrin is discontinued. The risk increases with increasing dose and with cumulative exposure. Due to the significant nature of visual loss, vigabatrin should be discontinued within 2 to 4 weeks of treatment initiation if no substantial improvement in seizure control is appreciated. For those who are continued on treatment, periodic ophthalmologic evaluation starting at 4 weeks and every 3 months thereafter is recommended. Reassessment should also be done 3 to 6 months after vigabatrin is discontinued. It should not be used in patients with or who have increased risk of other irreversible visual impairment. Due to the significant risk of visual loss, vigabatrin is only available through a restricted distribution program called SHARE.
Vigabatrin causes MRI abnormalities characterized by symmetric increased T2 signal and restricted diffusion involving the thalamus, basal ganglia, brainstem, and cerebellum. This can be seen in up to 22% to 32% of patients, but it tends to resolve when vigabatrin is discontinued.
Neurotoxicity also occurs with vigabatrin. Vacuolization due to accumulation of fluid resulting in the separation of the outer layer of myelin has been reported in laboratory animals at doses within the therapeutic range used in humans. This lesion is referred to as intramyelinic edema (IME). Similar to the MRI abnormalities described earlier, these lesions tend to resolve when vigabatrin is discontinued.
Drug Interaction
Vigabatrin can lower total plasma level of phenytoin by an average of 20%. This is due to induction of cytochrome P450 2C. Similarly, phenobarbital level is reduced on average by 8% to 16% and valproic acid by 8%. Although these may not be clinically significant, dose adjustment may be warranted if clinically indicated. Conversely, concomitant use of carbamazepine, clorazepate, primidone, and sodium valproate do not affect the plasma concentration of vigabatrin.
Use in Special Population
Dose adjustment of vigabatrin is recommended in patients with mild (CrCl 51–80 ml/min), moderate (CrCl 31–50 ml/min), and severe (CrCl 11–30 ml/min) renal impairment. The pharmacokinetics of vigabatrin has not been studied in patients with hepatic impairment. Although no studies involving sufficient number of patients aged 65 years and older, care should be taken when prescribing vigabatrin in this age group because of the potential coexisting impairment in renal function. Renal clearance of vigabatrin is 36% lower in healthy elderly subjects (>65 years) than in young healthy males. A single oral dose of 1.5 g of vigabatrin in elderly patients, greater than 65 years, with reduced creatinine clearance (<50 ml/min) can cause moderate to severe sedation and confusion, which can last up to 5 days.
Pediatric Use
The safety or the efficacy of vigabatrin in children (<16 years of age) with complex partial seizures has not been established. Animal studies have shown both neurobehavioral (convulsions, neuromotor impairment, and learning deficits) and neuropathological (brain vacuolation, decreased myelination, and retinal dysplasia) abnormalities in treated animals.
CLOBAZAM
Indication
Clobazam is a 1,5 benzodiazepine indicated as adjunctive therapy for seizures in Lennox-Gastaut syndrome (LGS) in patients 2 years and older.
Dosing
Clobazam is available in 5 mg, 10 mg, and 20 mg tablets. It should be administered in two divided doses except the 5 mg dose, which can be given once a day. Although the dosing is based on body weight, it should be individualized based on efficacy and tolerability (Table 28.3). The steady state of clobazam and its active metabolite is attained in 5 and 9 days, respectively, so the dose should be increased no faster than weekly. Clobazam can be taken with or without food. The tablets can be taken whole or crushed and mixed with applesauce.




