Toxic and Metabolic Disorders
Martin A. Samuels
Background
1. Hepatic encephalopathy describes the neurologic manifestations of liver failure.
2. The clinical categories of hepatic encephalopathy are
a. Acute hepatic encephalopathy
b. Chronic recurrent hepatic encephalopathy
c. Chronic progressive hepatic encephalopathy
1) Wilson disease
2) Acquired non-wilsonian hepatocerebral degeneration
Pathophysiology
1. The sine qua non for the development of hepatic encephalopathy is shunting of blood from the portal circulation to the systemic circulation with inadequate detoxification by a normally functioning liver.
2. This may occur because of endogenous liver disease (e.g., hepatitis, cirrhosis), portosystemic shunts (intrahepatic or extrahepatic), or both.
3. Clinical and experimental evidence support two potential mechanisms underlying the neurologic effects of portosystemic shunting of blood.
a. Endogenous benzodiazepine-like substances are incompletely metabolized by the liver and thus reach the brain and bind to γ-aminobutyric acid (GABA) receptors, resulting in excessive inhibition of neuronal function and thereby creating an encephalopathy. The benefit of the benzodiazepine antagonist, flumazenil, may relate to this mechanism.
b. Ammonia that is inadequately detoxified by the hepatic urea cycle reaches the brain where it is normally metabolized by the glutamate-glutamine system in astrocytes. When this system becomes saturated, ammonia reaches neurons where it is directly toxic, thereby producing the encephalopathy. The typical astrocytic changes seen in all forms of hepatic encephalopathy (Alzheimer type II gliosis) may be due to the upregulation of the glutamate-glutamine detoxification system in these cells. Improvement of hepatic encephalopathy with lowering of the serum ammonia concentration is taken as evidence for this mechanism.
Prognosis
1. Hepatic encephalopathy may be acute, recurrent, subacute, or chronic.
2. The prognosis depends on the underlying cause.
Diagnosis
1. The major clinical manifestations of hepatic encephalopathy are
a. Alteration of the level of consciousness including confusion with or without agitation (delirium), drowsiness, stupor, and coma.
b. Movement disorders, the most frequent of which are asterixis, myoclonus, and tremor.
c. Symptoms and signs of corticospinal tract disease are common (i.e., leg weakness, spasticity, increased tendon reflexes, and Babinski signs) and occasionally are the sole manifestation of hepatic encephalopathy (“hepatic paraplegia”).
d. Extrapyramidal symptoms and signs (i.e., rigidity, bradykinesia, and dysarthria) are common, particularly in the chronic hepatocerebral degenerations.
2. Hepatic encephalopathy should be considered in a patient with unexplained encephalopathy who might have liver disease, portosystemic shunting, or both. The diagnosis is more likely when
a. The blood ammonia is elevated. Arterial ammonia may be slightly better correlated with the clinical state than venous ammonia, but both are unreliable.
b. There is high-intensity signal in the basal ganglia (particularly the putamen and globus pallidus) on T1-weighted magnetic resonance imaging (MRI). This signal may represent deposition of paramagnetic substances (e.g., manganese and copper), although the reason for this deposition is unknown.
c. High-amplitude triphasic sharp waves are seen on the electroencephalogram (EEG). Although not specific for hepatic encephalopathy, the finding suggests a metabolic cause.
Treatment
1. Reduce sedating drugs to a minimum.
2. Correct fluid and electrolyte disturbances.
3. Treat concomitant infection.
4. Reduce protein load by
a. Searching for and treating any gastrointestinal (GI) hemorrhage.
b. Prescribing a low-protein diet, but providing enough calories to prevent proteolysis. Each liter of 10% dextrose in water provides 400 kcal. If nasogastric feeding is possible, 10% dextrose in water and lipids may be given to provide about 25 kcal/kg/d. The diet should be supplemented with vitamins (folate 1 mg/d, vitamin K 10 mg/d, and multivitamins).
c. Administering cathartics to help eliminate whatever protein remains in the bowel. Magnesium citrate 20 mL or sorbitol 50 g in 200 mL water may be administered via nasogastric tube or by mouth.
d. Administering lactulose (a synthetic disaccharide that cannot be digested in the upper GI tract) will allow large-bowel bacteria to metabolize the sugar, thus producing hydrogen ions that convert ammonia (NH3) to ammonium (NH4). Lactulose is not neurotoxic and is eliminated in the stool. Lactulose 30 to 50 mL (0.65 g/mL) may be administered by mouth, by nasogastric tube, or by retention enema three times a day.
5. Flumazenil (Romazicon) (0.2 mg/min intravenous [IV]) may have a temporary beneficial effect on the encephalopathy.
6. Hepatic transplantation may be lifesaving for patients with hepatic failure and reverses most of the neurologic manifestations of hepatic encephalopathy. In general, the longer the encephalopathy has been present, the lower the chance that it will be improved with liver transplantation.
Background
1. Hyperosmolality is defined as a serum osmolality of greater than 325 mOsm/L.
2. Osmolality may be measured directly or may be estimated using the formula:
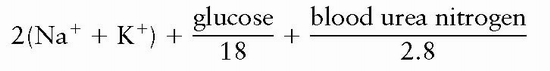
3. The difference between measured and calculated osmolality is the osmolal gap (normally < 10).
4. Effective osmolality is called tonicity. Substances that cross cell membranes freely (e.g., urea) may raise osmolality but have little or no effect on tonicity.
Pathophysiology
1. As can readily be appreciated from the determinants of osmolality, in most clinical settings, hyperosmolality is due to hypernatremia, hyperglycemia, azotemia, or the iatrogenic addition of extrinsic osmoles (e.g., alcohols, mannitol, glycerol).
2. Hypernatremia is defined as serum sodium (Na) concentration greater than 145 mEq/L. In all tissues other than the nervous system, hypernatremia leads to attraction of intracellular water, leading to cell shrinkage. The nervous system is unique in that it is capable of generating solute (e.g., idiogenic osmoles) such as glutamine and taurine to minimize cell shrinkage. When hypernatremia is prolonged or unusually severe (serum Na over 160 mEq/L) these mechanisms fail, leading to encephalopathy. When hypernatremia occurs, thirst increases and antidiuretic hormone (ADH) is released, leading to renal retention of pure water and thereby lowering serum Na toward normal. Hypernatremia is thus due to a defect in thirst, inadequate release or effect of ADH, loss of hypotonic fluid, or retention of Na.
3. Hyperglycemia is nearly always due to diabetes mellitus, caused either by inadequate insulin production or end-organ insulin resistance. In neurologic patients, this is often precipitated by the therapeutic use of glucocorticoids and some antiepileptic drugs such as phenytoin.
4. Azotemia is due to renal failure or inadequate renal perfusion (prerenal azotemia).
5. The ingestion of alcohols (e.g., ethanol, methanol) contributes to the osmolal gap, thereby increasing osmolality and tonicity.
6. Hyperosmolar agents such as mannitol or glycerol are often used in neurologic patients and may result in hyperosmolality.
7. Pertinent to almost all metabolic encephalopathies is the rate of change of the metabolite, slower elevations or depressions being better tolerated than acute ones.
Prognosis
1. Hyperosmolality usually produces a generalized encephalopathy without localizing or lateralizing features, but an underlying focal lesion (e.g., stroke, multiple sclerosis, neoplasm) may become symptomatic under the metabolic stress of a hyperosmolar state.
2. The prognosis of the hyperosmolality itself is good, but the long-term outlook depends on the cause.
3. For unknown reasons, hyperosmolality alone, particularly when due to hyperglycemia, may lead to continuous partial seizures, even when careful studies fail to uncover any underlying lesion. These seizures generally respond promptly to lowering of the serum glucose.
Diagnosis
1. The diagnosis is made by calculating the serum osmolality using the formula 2(Na+ K+) + glucose/18 + BUN/2.8 and by directly measuring osmolality using freezing point depression.
2. The difference between the measured and calculated osmolality is termed the “osmolal gap,” which should be less than 10 mOsm/L in normal circumstances.
3. An increased osmolal gap reflects the presence of solute such as alcohols, ethylene glycol, or therapeutic substances such as mannitol, sorbitol, or glycerol.
Treatment
1. Calculate the water losses using the following approach:
a. Calculate the normal total body water (NTBW) as follows: Body weight (in kilogram) × 0.6 = NTBW
b. Calculate the total body Na (TBS) as follows: NTBW × 140 mEq/L = TBS
c. Calculate the patient’s body water (PBW) as follows: TBS/patient’s serum Na = PBW
d. Calculate the patient’s water deficit (PWD) as follows:
NTBW – PBW = PWD
2. Replace the water losses so that the serum Na falls no faster than 2 mEq/L/hr using
a. Normal saline in hypovolemic patients (i.e., those with azotemia and/or hypotension).
b. Water in hypervolemic patients.
c. Renal dialysis if there is acute or chronic renal failure.
3. Insulin is administered (with frequent blood sugar testing) if there is hyperglycemia.
a. Intramuscular (IM) and subcutaneous insulin may be unpredictably absorbed, particularly in hypovolemic patients, because of poor tissue perfusion.
b. Rapid-acting insulin 0.1 U/kg by IV push followed by 0.05 U/kg/hr by continuous IV infusion is usually sufficient to reduce the blood sugar adequately and safely.
Background
Hyponatremia is defined as a serum Na of less than 125 mEq/L.
Pathophysiology
1. Hypotonicity is always associated with hyponatremia, but hyponatremia may be isotonic (e.g., infusion of salt-poor solutions, hyperlipidemia, or hyperproteinemia); hypertonic (e.g., hyperglycemia, mannitol); or hypotonic (impairment of free water excretion or an enormous free water load, as in psychogenic water drinking).
2. Tonicity (effective osmolality) is measured in the clinical laboratory. The difference between the calculated and measured osmolarity (the osmolal gap) should not exceed 10 mOsm/L (see section on treatment of hypernatremia above).
Prognosis
1. The prognosis of hyponatremia depends on the rate and magnitude of the fall in serum Na and its cause.
2. In acute hyponatremia (a few hours or less), seizures and severe cerebral edema may be rapidly life threatening at serum Na levels as high as 125 mEq/L, whereas patients may tolerate very low serum Na levels (even below 110 mEq/L) if the process develops slowly. Rapid correction of acute hyponatremia may be lifesaving, whereas rapid correction of chronic hyponatremia may be dangerous. Nervous system cells compensate for chronic hyponatremia by excreting solute to avoid water retention. If upon this substrate, serum Na rapidly rises, brain cells can rapidly shrink, causing osmotic demyelination (formerly known as central and extra pontine myelinolysis).
3. The cause of hypotonic hyponatremia is best determined by dividing all possibilities into three categories on the basis of the clinical estimate of the state of the extracellular fluid space. Blood pressure and heart rate with orthostatic measurements, the degree of engorgement of the neck veins, and the presence or absence of the third heart sound (S3) allow all patients with hypotonic hyponatremia to be categorized into three types:
a. Hypovolemic (reduced effective blood volume): hypotension, tachycardia with orthostatic worsening
b. Hypervolemic (edematous states)
c. Isovolemic (retention of free water)
Diagnosis
1. The diagnosis is made by measurements of the serum Na and serum osmolality, followed by an assessment of extracellular volume.
2. The major diagnoses in each category are
a. Hypertonic hyponatremia
1) Alcohols
2) Sugars
b. Isotonic hyponatremia (artifactual or pseudohyponatremia)
1) Lipids
2) Proteins
c. Hypotonic hyponatremia
1) Hypovolemic
a) Gastrointestinal Na losses
b) Hemorrhage
c) Renal salt wasting (including cerebral salt wasting syndrome)
d) Diuretic excess
e) Adrenal insufficiency
2) Hypervolemic
a) Congestive heart failure
b) Hepatic failure with ascites
c) Nephrotic syndrome
3) Isovolemic hyponatremia
a) Syndrome of inappropriate secretion of antidiuretic hormone (SIADH)
b) Psychogenic water drinking
c) Acute and chronic renal failure
d) Resetting of the osmostat (the sick cell syndrome)
Treatment
1. Hypertonic hyponatremia
a. Treat the underlying disorder (e.g., hyperglycemia, exposure to mannitol)
b. Replace only estimated salt losses
2. Isotonic hyponatremia
a. No fluid treatment for pseudohyponatremia disorders (e.g., hyperlipidemia, hyperproteinemia)
b. Reduce Na-poor solutions if possible (dextrose, mannitol)
3. Hypotonic hyponatremia
a. Hypovolemic hypotonic hyponatremia
1) Replace volume with isotonic saline
2) Treat underlying renal, adrenal, gastroenterologic conditions
3) Recognize and treat causes of cerebral salt wasting (e.g., intracerebral or subarachnoid hemorrhage)
b. Hypervolemic hypotonic hyponatremia
1) Free water restriction
2) Treat underlying edematous disorders (congestive heart failure, liver failure, nephrotic syndrome)
C. Isovolemic hypotonic hyponatremia
1) Chronic, slowly developing
a) Water restriction
b) Antagonize ADH with lithium, demeclocycline, or conivaptan if water restriction fails
2) Acute (less than 48 hours), rapidly developing
a) Three percent saline (containing 513 mEq/L of Na) 300 to 500 mL IV over 1 hour will correct at about 1 mEq/L/hr for 4 hours; then slow the correction rate to less than 10 mEq/L per 24 hours
b) Free water restriction or normal (0.9%) saline
Background
Hypokalemia is defined as a serum potassium level below 3.5 mEq/L.
Pathophysiology
1. Serum potassium may be low because of abnormal intracellular or extracellular potassium balance or because of excessive potassium losses (renal or extrarenal).
2. Hypokalemia due to excessive cellular potassium uptake may be due to
a. Insulin
b. Catecholamines
c. β2-Adrenergic agonists
d. Hypokalemic periodic paralysis
e. Alkalosis
f. Hypothermia
3. Extrarenal potassium loss (urine K+ less than 20 mEq/d) may be caused by
a. Diarrhea (low serum bicarbonate)
b. Cathartics; sweating (normal serum bicarbonate)
c. Vomiting (high serum bicarbonate)
4. Renal potassium loss (urine K+ more than 20 mEq/d) may be due to
a. Hyperreninemia
b. Hyperaldosteronism
c. Renal tubular acidosis
d. Diuretic use
e. Hypomagnesemia
f. Excessive glycyrrhizic acid (licorice) intake
Prognosis
Severe hypokalemia (serum potassium less than 1.5 mEq/L) may be life threatening due to cardiac arrhythmia and severe muscle weakness.
Diagnosis
1. The diagnosis of hypokalemia is made by serum potassium measurement.
2. Urinary potassium measurement may help determine whether the potassium loss is renal or extrarenal, but it should be borne in mind that such measurements are only valid in the face of a normal dietary and urinary Na, as Na restriction may result in some masking of renal potassium wastage.
3. The measured serum sodium bicarbonate, plasma renin, plasma aldosterone, urinary chloride levels, and blood pressure may also help in the differential diagnosis of the cause of hypokalemia.
4. The electrocardiogram usually shows a characteristic pattern consisting of U waves and a lengthened repolarization time (Q-U interval), a circumstance that predisposes to dangerous arrhythmias.
Treatment
1. Correct potassium balance problems, if possible (e.g., reduce β2-adrenergic agonists).
2. Dietary sodium restriction (less than 80 mEq/d) will reduce renal potassium losses.
3. Give oral potassium chloride (KCl) for mild hypokalemia (30 to 35 mEq/d).
4. For moderate (1.5 to 3.0 mEq/L) or severe (less than 1.5 mEq/L) hypokalemia, especially with cardiac arrhythmias and/or severe muscle weakness, IV KCl may be administered at the rate of 15 mEq over 15 minutes with continuous cardiac monitoring, aiming for a 1-mEq/L increase in the serum potassium. Thereafter, the rate should be slowed to less than 5 mEq/hr of a solution of KCl no more concentrated than 60 mEq/L.
Background
Hyperkalemia is defined as a serum potassium concentration of greater than 5 mEq/L.
Pathophysiology
1. Hyperkalemia may be seen in circumstances that may or may not cause an excess of whole-body potassium.
2. The common causes of hyperkalemia without an excess of potassium are
a. Muscle injury (e.g., trauma, persistent seizures, muscle infarction)
b. β2-Adrenergic antagonists (e.g., propranolol)
c. Insulin resistance
d. Metabolic acidosis
e. Digitalis poisoning
f. Depolarizing muscle relaxants (e.g., succinylcholine)
g. Hyperkalemic periodic paralysis (muscle sodium channel mutation)
3. Common causes of hyperkalemia caused by whole-body potassium excess include
a. Addison disease
b. Aldosterone deficiency or antagonism (e.g., hyporeninemia; angiotensinconverting enzyme [ACE] inhibitor therapy; nonsteroidal anti-inflammatory drugs [NSAIDs]; heparin)
c. Aldosterone resistance (e.g., renal failure, renal tubular disorders, potassiumsparing diuretics)
Prognosis
1. The prognosis of hyperkalemia depends upon its effects on the electrocardiogram (ECG) and muscle membranes.
2. The first sign of hyperkalemia is usually peaking of the T wave of the ECG, which usually occurs with a potassium level of about 6.0 mEq/L. As the potassium rises, the QRS complex widens, followed by reduction in its amplitude and then disappearance of the T wave.
3. Muscle weakness usually develops when the potassium is greater than 8 mEq/L.
Diagnosis
1. Hyperkalemia is suspected when the characteristic ECG pattern is seen, particularly when combined with weakness and sometimes, with paresthesias.
2. The diagnosis is confirmed with measurement of the serum potassium.
Treatment
1. If hyperkalemia is considered life threatening because it is producing ECG changes and/or severe muscle weakness, one should treat by protecting the heart against life-threatening arrhythmias, promoting redistribution of potassium into cells, and enhancing potassium removal.
2. Cardiac protection: calcium gluconate 10% solution, 20 mL by rapid IV infusion
3. Redistribution into cells
a. Glucose 50 g/hr IV
b. Insulin 5 units IV push every 15 minutes
c. Albuterol 10 to 20 mg by inhaler
4. Enhance removal of potassium
a. Sodium polystyrene sulfonate (Kayexalate) 15 to 60 g with sorbitol by mouth (p.o.) or 50 to 100 g with retention enema
b. Hemodialysis
c. Loop diuretics
1) Furosemide 40 to 240 mg IV over 30 minutes
2) Ethacrynic acid 50 to 100 mg IV over 30 minutes
3) Bumetanide 1 to 8 mg IV over 30 minutes
Background
1. Neurologic syndromes appear with serum concentrations of calcium above 12 mg/dL if serum albumin is normal. With low serum albumin, ionized calcium is higher and neurologic manifestations appear at lower electrolyte levels.
2. Anorexia, constipation, nausea, fatigue, and headache are early features. At higher levels of calcium, confusion, coma, rigidity, and myoclonus occur. Convulsions are rare.
Pathophysiology
1. In younger individuals, the most common cause is hyperparathyroidism and in older persons, it is bone tumors including widespread metastases and multiple myeloma. Excess intake of vitamin D, sarcoidosis, and prolonged immobilization are less frequent causes of hypercalcemia.
Prognosis
1. All features are reversible unless there has been respiratory arrest.
Diagnosis
1. Determination of serum concentrations of calcium and albumin is required.
2. Parathyroid hormone levels and evaluation for the above underlying diseases (bone imaging, chest X-ray, serum protein electrophoresis immunoelectrophoresis) are required if there is no apparent explanation for the syndrome.
3. The QT interval is often shortened.
Treatment
1. Hydration with normal saline at high rates of infusion is the primary treatment. Four L of IV fluids per 24 hours is appropriate if there is no congestive heart failure.
2. For severe symptoms with Ca++ level over 12 mg/dL, calcitonin is given, 4 to 8 U/kg subcutaneously every 6 to 12 hours. This is rapidly effective.
3. Pamidronate is more slow acting (3 to 5 days) but with prolonged effect. Doses range from 30 to 60 mg for calcium levels of 12 to 14 mg/dL to 90 mg for calcium levels over 16 mg/dL. The drug is infused slowly, over about 3 hours, in 300 mL normal saline.
Vitamin A
Background
1. Vitamin A deficiency is an important cause of blindness in large parts of the world but is rare in economically developed countries.
2. Vitamin A intoxication is seen in people who engage in megavitamin therapy or who have ingested large amounts of animal tissue that concentrates vitamin A (e.g., bear liver).
Pathophysiology
In many developing countries, general malnutrition is the major cause of vitamin A deficiency, whereas in developed countries it is usually related to malabsorption or an unconventional diet.
Prognosis
1. If treated early, the neurologic manifestations are usually completely reversible.
2. Once blindness has occurred, little can be done to reverse the visual loss.
Diagnosis
1. Night blindness and dry eyes are probably the earliest symptoms of vitamin A deficiency.
2. Dry pruritic skin is also an early symptom of this deficiency.
3. Hypervitaminosis A may cause the syndrome of pseudotumor cerebri.
Treatment
1. Vitamin A 1,000 units daily for 6 months should be given and a normal diet should be restored.
2. Vitamin A up to 100,000 units daily for 6 months with restoration of a normal diet may be needed for moderate or advanced symptoms.
3. Long-term use of vitamin A is not advisable as it may produce hypercoagulable state with consequent increased intracranial pressure (ICP) (pseudotumor cerebri) possibly caused by cerebral venous thrombosis. Treatment for this consists of discontinuation of vitamin A.
Vitamin B1 (Thiamine) Deficiency
Background
1. Vitamin B1 (thiamine) deficiency occurs in parts of the world where polished rice is a major dietary staple or in people who are malnourished for any reason.
2. In developed countries, it is strongly linked to alcoholism.
Pathophysiology
Thiamine is the coenzyme in thiamine pyrophosphate catalysis of decarboxylation of pyruvic acid and α-ketoglutaric acid.
Prognosis
Treatment of Wernicke encephalopathy (the central nervous system [CNS] disease caused by thiamine deficiency) is usually quite successful, but the longer treatment is delayed, the greater the probability of irreversible brain disease. (See later section.)
Diagnosis
1. Thiamine deficiency should be assumed to be present in all malnourished people including, but not limited to, those with alcoholism.
2. The full triad of Wernicke encephalopathy (i.e., mental change, ataxia, and oculomotor findings) is present in only a minority of those people later found to have Wernicke encephalopathy by pathologic study.
3. Measurement of 24-hour urine thiamine excretion is available, and red blood cell transketolase may be measured.
4. As confirmation of the diagnosis, lesions characteristic of Wernicke encephalopathy (i.e., small mamillary bodies and/or hypothalamic peri-third ventricular necrosis) may be seen on MRI.
Treatment
1. Thiamine 100 mg by rapid IV infusion followed by
2. Thiamine 25 mg daily for several months and restoration of a normal diet
Vitamin B2 (Riboflavin) Deficiency
Background
Riboflavin deficiency is caused by general malnutrition or malabsorption.
Pathophysiology
Riboflavin is a coenzyme in the flavoprotein enzyme system.
Prognosis
Treatment is usually successful unless the disease is far advanced.
Diagnosis
1. The clinical syndrome of cheilosis, angular stomatitis, visual loss, night blindness, glossitis, and burning feet in a susceptible person suggests the diagnosis.
2. Twenty-four-hour urinary riboflavin excretion measurements are available (less than 50 μg per 24 hours indicates the deficiency) but are rarely used except in problematic diagnostic dilemmas.
Treatment
1. Riboflavin 5 mg p.o. three times a day (t.i.d.)
2. Vitamin A replacement may help in relieving riboflavin-induced ocular symptoms (see Treatment section of Vitamin A, above)
3. Restoration of a normal diet
Niacin (Nicotinic Acid, Nicotinamide, B3) Deficiency
Background
Niacin deficiency (pellagra) is usually associated with general malnutrition and often with alcoholism.
Pathophysiology
Niacin is the coenzyme for nicotinamide dinucleotide codehydrogenase for the metabolism of alcohol, lactate, and L-hydroxybutyrate.
Prognosis
Untreated pellagra is lethal, but if recognized during life, it will usually respond favorably to therapy.
Diagnosis
1. The characteristic triad of dermatitis (sun sensitivity with scaling eruption followed by hyperpigmentation), diarrhea, and mental symptoms (usually a disorder of attention and/or mood followed by confusion, drowsiness, stupor, and coma) suggests the diagnosis in the setting of malnutrition.
2. The diagnosis can be confirmed with a 24-hour urinary niacin excretion of less than 3 mg per 24 hours.
Treatment
1. Niacin or nicotinamide 50 mg p.o. ten times daily for 3 weeks
2. In patients unable to take oral feedings, nicotinamide may be given IV 100 mg/d for 5 to 7 days
3. Resumption of a normal diet is important for long-term recovery
4. If pyridoxine deficiency is also deemed to be present (e.g., isoniazid therapy), vitamin B6 (pyridoxine) must also be replaced as it is required for the normal conversion of tryptophan to niacin
Vitamin B6 (Pyridoxine) Deficiency, Dependency, and Toxicity
Background
1. Pyridoxine deficiency is rarely seen in developed countries except in people who are taking isoniazid, an antituberculosis drug that is an antagonist of pyridoxine.
2. Cycloserine, hydralazine, and penicillamine also may lead to pyridoxine deficiency.
3. Pyridoxine toxicity is seen in people who take more than the recommended daily allowance of 2 mg because of perceived health benefits of megavitamin therapy.
Pathophysiology
Pyridoxine is a cofactor in the conversion of tryptophan to 5-hydroxytryptophan and the conversion of homocysteine to cystathionine.
Prognosis
Treatment usually results in complete resolution of the complaints.
Diagnosis
1. Pyridoxine deficiency causes a generalized sensory and motor neuropathy.
2. Pyridoxine dependency is a rare autosomal recessive condition that leads to neonatal seizures.
3. Pyridoxine overuse also causes a peripheral neuropathy.
a. Long-term low-dose (about 50 mg/d) exposure to pyridoxine leads to a smallfiber neuropathy.
b. Shorter exposure to very high doses (over 100 mg/d) may produce a primary sensory neuronopathy that is less likely to improve with cessation of exposure to the vitamin.
Treatment
1. For pyridoxine deficiency caused by
a. Malnutrition: 50 mg/d p.o. for several weeks followed by 2 mg/d and resumption of a normal diet
b. Pyridoxine antagonists: 50 mg/d only while taking the antagonist
2. For pyridoxine dependency: 10 mg by rapid IV infusion to terminate neonatal seizures and then 75 mg/d for life
3. Pyridoxine toxicity: discontinue pyridoxine supplementation
Vitamin B12 (Cobalamin) Deficiency
Background
1. Vitamin B12 deficiency may result from inadequate dietary intake, but this is infrequent as the daily requirement is small (2 μg/d) and the body stores are high (4 mg or about a 7-year supply).
2. Vegans who assiduously avoid animal protein may become cobalamin-deficient, but this process requires many years.
3. Normal salivary amylase is required to separate cobalamin from food. In rare circumstances (e.g., Sjögren syndrome), salivary amylase deficiency may cause cobalamin deficiency.
4. More commonly, cobalamin deficiency is caused by failure to mobilize vitamin B12 from the GI tract because of insufficient intrinsic factor, most often caused by autoimmune gastritis (pernicious anemia).
5. Aging alone may lead to enough gastric parietal cell atrophy to cause intrinsic factor deficiency and consequent vitamin B12 deficiency.
6. In rare circumstances, the ingested cobalamin may be consumed before absorption by a parasite (the fish tapeworm Diphyllobothrium latum) or may be inaccessible to cells because of a genetically determined deficiency in one of the cobalamin-carrying proteins (transcobalamin I and II).
7. Human immunodeficiency virus (HIV) infection may lead to abnormal cobalamin function by an unknown mechanism, possibly involving abnormal transmethylation. This may explain why the pathology of HIV-induced spongiform myelopathy is so similar to that of the myelopathy caused by cobalamin deficiency.
Pathophysiology
1. Cobalamin is bound to salivary R protein. In the duodenum, pancreatic enzymes digest the R protein allowing cobalamin to be bound to intrinsic factor that is synthesized in gastric parietal cells. The cobalamin-intrinsic factor dimer is absorbed by specific receptors in the microvilli of the distal ileum. The newly absorbed cobalamin enters the portal circulation bound to transcobalamin II. Transcobalamin I is bound to previously absorbed cobalamin.
2. Inside cells, cobalamin is converted to its two active forms, methylcobalamin and adenosylcobalamin.
a. Methylcobalamin is the coenzyme for the enzyme methionine synthetase (also known as methyltransferase), which catalyzes the conversion of homocysteine to methionine. Cobalamin is then remethylated to methylcobalamin by a methyl group donated by methyltetrahydrofolate (serum folate). By this process, the demethylated folate may participate in the formation of thymidylate, which is required for DNA synthesis. These interlocking reactions account for the fact that many of the clinical manifestations of vitamin B12 and folate deficiencies are similar.
b. Cobalamin also participates in an important metabolic pathway that is independent of folate. In mitochondria, adenosylcobalamin acts as a coenzyme for methylmalonyl-coenzyme A (CoA) mutase, which catalyzes the conversion of methylmalonyl-CoA to succinyl-CoA. Thus, homocysteine and methylmalonic acid act as biologic markers for the intracellular effectiveness of cobalamin’s two coenzymes.
Prognosis
1. The clinical features of the cobalamin deficiency syndrome are predominantly demyelination of the lateral and posterior columns of the spinal cord (subacute combined degeneration), the white matter of the brain, and of the optic nerves. A peripheral neuropathy may also be present.
2. Patients usually present with upper extremity paresthesias followed by stiffness of the legs, slowness of thinking, and reduced visual acuity. For unknown reasons,
the optic neuropathy or mental change may dominate the clinical picture in some patients.
3. Most of the manifestations of the disease are reversible with appropriate therapy, but advanced disease may not completely respond.
4. Exposure to nitrous oxide may precipitate an acute presentation of cobalamin deficiency (anesthesia paresthetica), as it is an inhibitor of methyltransferase, one of the enzymes for which cobalamin is a coenzyme.
Diagnosis
1. Hypersegmented (i.e., greater than five lobes) polymorphonuclear leukocytes are often seen on the peripheral blood smear.
2. Bone marrow may show megaloblasts (i.e., red blood cell precursors with a relatively immature nucleus compared to the cytoplasm).
3. Vitamin B12 levels are usually low
a. When less then 100 pg/mL, cobalamin deficiency is likely.
b. When between 100 and 180 pg/mL, cobalamin deficiency is possible.
c. When over 180 pg/mL, cobalamin deficiency is unlikely.
4. Serum methylmalonic acid is the most specific test for intracellular cobalamin failure. Levels above 0.5 μmol/L suggest intracellular cobalamin failure.
5. The Schilling test may be useful to determine the cause of vitamin B12 deficiency.
a. Phase I is aimed at determining whether the patient can absorb crystalline vitamin B12.
b. Phase II identifies those who are vitamin B12-deficient because of intrinsic factor deficiency.
c. The phase III Schilling test, in which radiolabeled vitamin B12 is attached to albumin, is used to identify those patients who are unable to extract vitamin B12 from food because of an inadequately acidic environment.
6. Anti-intrinsic factor antibodies are specific but insensitive for autoimmune gastritis.
7. Anti-parietal cell antibodies are sensitive but not specific for autoimmune gastritis.
Treatment
1. Cyanocobalamin 1,000 μg IM daily for 1 week, followed by weekly injections for 1 month, followed by monthly injections for life
2. Cyanocobalamin 1 mg/d p.o. may be effective, particularly in elderly patients with gastric atrophy. Methylmalonic acid levels should be monitored to ensure that the treatment is having the expected metabolic effect
3. Discontinue exposure to nitrous oxide
Vitamin B9 (Folate) Deficiency
Background
1. Folate is synthesized by plants and microorganisms. Its major dietary source is green, leafy vegetables.
2. The daily requirement is 50 μg except in pregnant and lactating women, for whom it is increased approximately 10-fold.
3. Folate is ingested as a polyglutamate, which is metabolized to pteroylmonoglutamate and absorbed in the jejunum. In the bowel mucosal cells, it is reduced to tetrahydrofolate and methylated to methyltetrahydrofolate (serum folate).
4. Only about a 12-week supply of folate is stored in the body, so folate deficiency may become rapidly evident with malnutrition.
Pathophysiology
1. Folate interacts intimately with vitamin B12 (cobalamin). Serum folate (methyltetrahydrofolate) is the methyl donor that reconstitutes cobalamin into methylcobalamin in the conversion of homocysteine to methionine. Thus, a reduction in
homocysteine levels are a reflection of the effectiveness of both folate and vitamin B12 in the methyltransferase (methionine synthetase) reaction.
2. Once demethylated, tetrahydrofolate undergoes polyglutamation and is converted to 5,10-methylene tetrahydrofolate, which, catalyzed by thymidylate synthase, generates deoxythymidine monophosphate for the synthesis of the thymidine needed for DNA synthesis.
3. Vitamin B12 deficiency causes release of folate from cells and interferes with its utilization, leading to an elevated serum folate level (the folate trap).
4. When vitamin B12 is repleted, the folate level may fall precipitously, leading to a folate-deficiency state unmasked by the cobalamin therapy.
Prognosis
1. Pure folate deficiency is rare as it is usually associated with generalized malnutrition, but it may be seen when folate inhibitors have been administered (e.g., methotrexate and sulfonamides are inhibitors of dihydrofolate reductase and phenytoin interferes with folate absorption).
2. Folate deficiency during gestation is associated with neural tube defects.
3. In adults, pure folate deficiency probably causes a sensory-motor length-dependent polyneuropathy. In most cases, folate repletion leads to reversal of the neurologic deficits and adequate provision of folate during pregnancy reduces the risk of neural tube defects.
Diagnosis
1. The blood and bone marrow changes of folate deficiency are indistinguishable from those caused by vitamin B12 deficiency.
2. A low serum folate level is specific but not particularly sensitive.
3. If the serum folate level is normal, but folate deficiency is suspected on clinical grounds, a red blood cell folate level should be obtained because it reflects the average intracellular folate level over the life span of the red blood cell and therefore is not unduly affected by recent dietary intake.
Treatment
1. Folic acid 1 mg/d p.o.
2. Resumption of a normal diet
3. For patients on folate antagonists, folinic acid (leucovorin, citrovorum factor) 15 mg p.o. is given every 6 hours for 10 doses starting 24 hours after the dose of methotrexate. If folate deficiency develops from phenytoin, another antiepileptic drug should be chosen because folate replacement may reduce the antiepileptic efficacy of phenytoin.
4. In pregnant women, daily folic acid 400 μg supplementation is recommended. For women with a history of neural tube defects, the daily recommended dose is 4 mg. In those women who take the larger dose, it should be administered as a dedicated folic acid capsule and not by taking additional multivitamin capsules, as that practice may lead to toxicity from other vitamins, particularly vitamin A (see Vitamin A intoxication above).
Vitamin C (Ascorbic Acid) Deficiency
Background
Vitamin C deficiency (scurvy) is rare in developed countries, occurring almost exclusively in generally malnourished people who are poor, elderly, alcoholic, or adherents to unusual diets.
Pathology
1. Ascorbic acid is found in citrus fruits, green vegetables, and tomatoes, and is absorbed from the small intestine via a transport system.
2. It has multiple functions, including acting as an antioxidant, a promoter of iron absorption, and a cofactor in the conversion of dopamine to norepinephrine and the synthesis of carnitine.
3. Consuming less than 10 mg of ascorbic acid daily will result in deficiency in a few months.
Prognosis
1. Vitamin C deficiency is characterized by symptoms and signs of abnormal connective tissue such as perifollicular hemorrhages and bleeding from the gums. Neurologic symptoms include weakness, fatigue, depression, and confusion.
2. Treatment usually results in complete remission of the clinical syndrome.
3. “Megadoses” of vitamin C (i.e., greater than 2 g/d) may result in GI bleeding and oxalate kidney stones, but no hypervitaminosis C syndrome of the nervous system is known.
Diagnosis
A vitamin C plasma concentration of less than 11 μmmol/L is considered abnormal, but most patients with scurvy and neurologic impairment have an undetectable plasma vitamin C level.
Treatment
Vitamin C 100 mg q.i.d. for 1 week, followed by 100 mg t.i.d. for 1 month and resumption of a normal diet.
Vitamin D Deficiency
Background
1. Vitamin D (1,25-dihydroxycholecalciferal; vitamin D3) is the least typical of the vitamins in that it can be synthesized in the skin in amounts adequate for metabolic needs provided there is adequate sun exposure.
2. Vitamin D deficiency or resistance is the cause of rickets in the growing skeleton and osteomalacia in adults.
Pathophysiology
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


