Fig. 1
Key components of the ascending arousal system. Cholinergic (ACh) inputs from the pedunculopontine (PPT) and laterodorsal tegmental (LDT) nuclei to the relay and reticular nuclei of the thalamus (yellow pathway) facilitate thalamocortical transmission. Monoaminergic cell groups, including the tuberomammillary nucleus (TMN) containing histamine (His), the A10 cell groups in ventral periaqueductal gray (vPAG) nucleus containing dopamine (DA), the median raphe nuclei containing serotonin (5–HT), and the locus coeruleus (LC) containing noradrenaline (NE) activate the cerebral cortex through a second pathway (red) to facilitate processing of inputs from the thalamus. This ventral pathway also receives contributions from peptidergic neurons in the lateral hypothalamus (LHA) containing orexin or melanin-concentrating hormone (MCH) and from basal forebrain (BF) neurons that contain gamma-aminobutyric acid (GABA) or ACh. Reprinted by permission from Macmillan Publisher Ltd: [Nature Publishing Group] (Saper et al. 2005), copyright (2005)
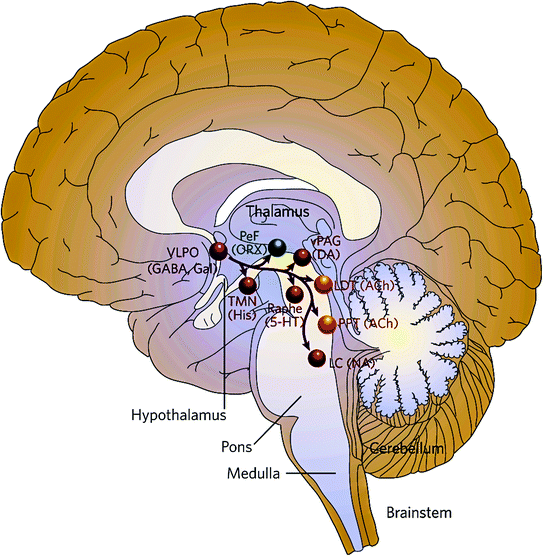
Fig. 2
Key projections of the ventrolateral preoptic nucleus (VLPO) to the main components of the ascending arousal system. The VLPO projects to the monoaminergic cell groups (red), such as the A10 cell groups in ventral periaqueductal gray (vPAG) nucleus containing dopamine (DA), the raphe cell groups, the tuberomammillary nucleus (TMN), and the locus coeruleus (LC). The VLPO also innervates neurons located in the lateral hypothalamus (LHA; green), including the perifornical (PeF) orexin (ORX) neurons, and interneurons in the cholinergic (ACh) cell groups (yellow), the pedunculopontine (PPT), and laterodorsal tegmental nuclei (LDT). 5–HT serotonin, GABA gamma-aminobutyric acid, gal galanin, NA noradrenaline, and His histamine. Reprinted by permission from Macmillan Publisher Ltd: [Nature Publishing Group] (Saper et al. 2005), copyright (2005)
3 Insomnia and Its Pharmacological Treatments
Thirty to fifty percent of adults complain of insomnia, which may be transient or episodic, short-term, or chronic in duration. Chronic insomnia impairs mood, motor performance, and cognition. Insomnia also is a common problem in children. The estimated prevalence of pediatric insomnia including bedtime refusal and night awakenings is 10–30 % among normal infants, toddlers, and preschoolers; in children with neurodevelopmental and psychiatric disorders, the prevalence rises to 50–75 % (Mindell et al. 2006). Chronic insomnia occurs in 4–7 % of children (Zhang et al. 2011).
3.1 Insomnia, Brain Morphology, and Cognitive Function
Chronic insomnia affects neuroplasticity and cognitive function. Functional magnetic resonance imaging (fMRI) scanning while performing a category and a letter fluency task demonstrated hypoactivation of the medial and inferior prefrontal cortical areas in chronic insomnia patients compared to normal controls, which recovered after 6 weeks of non-pharmacological sleep therapy (Altena et al. 2008). Chronic primary insomnia patients had reduced gray matter volume in the left orbitofrontal cortex, which strongly correlated with insomnia severity; gray matter volume in the anterior and posterior precuneus was also reduced (Altena et al. 2010). A transcranial magnetic stimulation (TMS) study in chronic insomnia patients found relatively reduced intracortical facilitation, but globally increased absolute excitability to both suprathreshold single- and paired-pulse stimulation, which persisted even after sleep therapy (Van der Werf et al. 2010). These findings favor the “activating” profile in chronic insomnia (Lanza et al. 2014) and could also suggest an optimal therapeutic window to start therapy before structural changes occur (Van der Werf et al. 2010). Further research is needed to elucidate this.
3.2 Insomnia Treatment
The primary treatment goals for insomnia are to improve sleep quality and quantity and to eradicate related daytime impairments (Schutte-Rodin et al. 2008). Psychological and behavioral interventions are recommended as initial interventions; they may also supplement hypnotic prescriptions (Schutte-Rodin et al. 2008).
Most hypnotic agents are approved for short-term use only. Some patients with severe or refractory insomnia or chronic comorbid illness use chronic hypnotic medications either nightly or intermittently, as needed. Adequate cognitive behavioral treatment (CBT) should be tried in these patients. Combined CBT and pharmacotherapy does not show any consistent advantage or disadvantage compared to CBT alone in patients with chronic insomnia.
Hypnotics presumably act either by blocking neurotransmitters that promote arousal or facilitating the neurotransmitters that drive sleep. Hypnotic agents (benzodiazepines and non-benzodiazepines) facilitate GABA receptors. Most of the agents that block wake promotion are histamine antagonists (usually H1 receptor antagonists, such as diphenhydramine, low-dose doxepin, low-dose mirtazapine, novel selective H1 receptor antagonists, and 5 HT2A antagonists); some of these agents may also exert antimuscarinic actions and/or anti-adrenergic effects. Orexin receptor antagonists also produce sleepiness with much greater and longer effects in man than any previous compound.
In children less than 5 years old, the mainstay of treatment remains empirically supported behavioral interventions (graduated extinction, parent education, delayed bedtime with removal from bed/positive bedtime routines) (Morgenthaler et al. 2006). Optimizing the sleep environment, sleep–wake scheduling, sleep practices, and addressing physiological factors (such as caffeine intake) are important adjunctive measures. The use of medications to treat pediatric insomnia should be approached carefully. Many forms of neuroplasticity peak during early developmental stages and might be affected by pharmacologic agents; injury to the developing brain can alter neuronal activity, modify synaptic mechanisms, and interfere with normal development and plasticity (Cramer et al. 2011). Pharmacological treatment is merely adjunctive and should follow general guidelines: (1) use the lowest effective dose and the shortest duration possible (usually <1 month), (2) avoid early evening dosing (due to circadian alerting), and (3) adjust dose based on pediatric metabolic profile of the drug. Comorbid primary sleep disorders should be addressed, and medication side effects should be monitored (Owens and Mindell 2011).
3.2.1 Benzodiazepine Receptor Agonists
GABA is the predominant inhibitory neurotransmitter in the central nervous system. The GABAA-benzodiazepine receptor complex is a ligand-gated ion channel complex, which consists of 3 moieties: a GABAA recognition site, a benzodiazepine recognition site, and a chloride ionophore. GABAA has at least 5 glycoprotein subunits, each of which include multiple isoforms (α1-6, β1-3, γ1-3, δ1, ε1, θ1, and ρ1-3) (Vinkers and Olivier 2012). The most common isoforms noted are Type I (α1β2γ2) and Type II (α3β2γ2) configurations.
GABA receptors mediate two forms of inhibition: Phasic inhibition is generated by postsynaptic GABAA receptors, while tonic inhibition is primarily mediated by extrasynaptic GABAA receptors that contain either the δ subunit or the α sub-unit (Whissell et al. 2013). GABAergic synapses are also capable of activity-dependent long-term plasticity and LTP/long-term depression and spike-timing dependent plasticity (Maffei 2011).
Various hypnotic medications target GABAA receptors. Increased GABAA-mediated inhibition in the thalamus and neocortex promotes slow oscillations and EEG spindles.
Classic benzodiazepines have a high affinity for multiple receptor subunits and bind to both Type I and Type II receptors, while non-benzodiazepine hypnotics, such as zolpidem and zaleplon, have more selective affinity for Type I receptors. Zolpidem’s affinity is highest for the α1βχγ2, while eszopiclone’s affinity is for α3 ≫ α1. Gaboxadol has a high affinity for extrasynaptic GABAA receptors δ subunit.
Absorption of benzodiazepine receptor agonists permits rapid sleep onset; the elimination half-life and dose determine the duration of somnogenic action and residual sedation (if any). Most benzodiazepines used as hypnotics (except for temazepam) have maximal absorption usually within 1–1.5 h; many are lipophilic and elimination half-lives vary.
The “Z” drugs (zolpidem, zaleplon, zopiclone, eszopiclone) have no significant effect on sleep-disordered breathing. The incidence of misuse of these agents is lower, although cases of chronic abuse, tolerance, misuse, and withdrawal symptoms of zolpidem have been reported (Victorri-Vigneau et al. 2007).
Adverse effects should be recognized. Cognitive impairment and impaired psychomotor performance can occur at peak levels, while long medication half-life can result in daytime sedation. Anterograde amnesia may occur with triazolam and the non-benzodiazepines, particularly when combined with alcohol. Confusional arousals or sleepwalking episodes or sleep eating have been reported. Nighttime falls in the elderly are increased. Other side effects include tolerance, abuse, and withdrawal. Rebound insomnia (the occurrence of insomnia symptoms worse than baseline) can occur for 1–2 days after abrupt withdrawal following prolonged benzodiazepine use.
Anterograde amnesia and impaired cognitive function may be related to changes in neuroplasticity with hypnotic use. GABAergic inhibition modulates excitatory synaptic plasticity, while GABA receptor blockers promote LTP of glutamatergic transmission in the hippocampus and neocortex (Nitsche et al. 2012). During high-frequency synaptic transmission, GABAergic inhibition is released by auto-inhibition of GABAergic terminals via GABAB receptors. This then allows postsynaptic depolarization to activate N-methyl-D-aspartate receptors and induce LTP (Davies et al. 1991). In rats, lorazepam (a long-acting benzodiazepine) impaired both recognition memory and synaptic plastic processes (long-term depression and LTP) in the peri-rhinal cortex (Wan et al. 2004). In cats, zolpidem significantly reduced cortical plasticity by ~50 %; this effect was not due to abnormal sleep architecture (Seibt et al. 2008). Also, in cats, triazolam (a short-acting benzodiazepine) impaired sleep architecture and resulted in EEG abnormalities, but did not affect cortical plasticity (Seibt et al. 2008). In mice, acute administration of δGABAA receptor agonist impaired memory behaviors and inhibited synaptic plasticity (Whissell et al. 2013).
The long-term effects of chronic use of benzodiazepines and benzodiazepine receptor agonists on neuroplasticity and cognitive function are of concern (Barker et al. 2004, 2005). Results from a meta-analysis of 13 studies that utilized psychological tests indicated that chronic insomnia patients on long-term benzodiazepine treatment had worse cognition compared to control subjects; impairment in certain domains persisted even after discontinuation of hypnotic use (Barker et al. 2004). Test performance in all 12 cognitive domains examined was worse in the benzodiazepine group, with effect sizes ranging in magnitude from −1.30 to −0.42 (Barker et al. 2004). Even after >6 months of withdrawal from long-term benzodiazepine use (mean use 108 months), verbal memory, motor control/performance, and nonverbal memory were impaired, while visuospatial skills and attention/concentration were not (Barker et al. 2004). Majority of the patients in the studies included in the above meta-analyses had utilized benzodiazepines to control anxiety/depression. Of the 7 studies that used a control group, 1 study used a healthy control group and an anxious control group, while 6 recruited control subjects from the general population with no history of anxiety (Barker et al. 2004). A caveat, therefore, in interpreting these results is that reduced performance after benzodiazepine withdrawal could be due to recurrence of pre-morbid symptomatology. Similar findings were reported in a study of 89 older adults (mean age ≥55 years) on chronic benzodiazepine therapy and who were studied at baseline and after withdrawal from benzodiazepines/benzodiazepine receptor agonists for a period up to 6 months. Prolonged impairment of attentional and psychomotor cognition function was noted; again, findings persisted for at least 6 months following drug withdrawal (Puustinen et al. 2014).
3.2.2 Antihistamines and Sedating Antidepressants
Histamine H1 receptors are present in the cortex, hippocampus, amygdala, hypothalamus, striatum, and cerebellum, while H2 receptors are located in the basal ganglia, amygdala, hippocampus, and cortex. The H3 receptors are found in the histaminergic neurons. Histamine receptors (H1 and H2) are postsynaptic, while H3 receptors are presynaptic. H1 receptor binding promotes wakefulness, while H2 receptor antagonists promote slow-wave sleep (Stahl 2008). H3 receptors are both autoreceptors and heteroreceptors: Binding at these sites inhibits histamine and norepinephrine release.
Activation of H1 or H2 receptors leads to downstream signaling cascades that can mediate functional and structural changes in synapses. H3 receptors might affect synaptic plasticity in the hippocampus and striatum by reducing the release of several neurotransmitters including glutamate. H3 receptor activation hinders synaptic transmission and reduces paired-pulse facilitation in the dentate gyrus (Kohler et al. 2011). Infusion of H1 and H2 receptor antagonists and H3 receptor agonist in the hippocampus of rats blocked long-term memory retention, suggesting that object recognition memory is modulated through H1, H2, and H3 receptors (da Silveira et al. 2013). An experiment in mice suggested that histamine-induced emotional memory consolidation is mediated by H1 but not H2 receptors (Gianlorenco et al. 2012).
Diphenhydramine blocks H1 receptors and is FDA-approved for short-term use to treat occasional sleeplessness. Doses in adults range from 50–100 mg at bedtime. Pediatric dosing ranges from 1 mg/kg up to 50 mg per day. Absorption is rapid, peak sedative effect occurs at 1–3 h after dose, and sedation lasts 4–7 h. Side effects include daytime drowsiness, cholinergic effects, and paradoxical excitation. Adult patients with rhinitis, taking 50 mg of diphenhydramine were more sleepy, less vigilant, and performed worse across all cognitive domains in a comprehensive battery of automated neuropsychological tests compared to patients on desloratadine 5 mg or placebo (Wilken et al. 2003).
Low-dose doxepin (Silenor) is FDA-approved for the treatment of sleep maintenance insomnia. Compared to placebo, low-dose doxepin (1, 3, 6 mg) significantly improved wake after sleep onset (WASO), total sleep time (TST), and overall sleep efficiency; at 6 mg, it also significantly reduced subjective latency to sleep onset (Roth et al. 2007). Doxepin is a tricyclic drug that blocks histaminergic, cholinergic, and alpha-1 noradrenergic receptors and also inhibits reuptake of norepinephrine and serotonin. Ultra-low-dose doxepin is a selective H1 antagonist, as it has high affinity for H1 receptors but little effect on serotonergic or adrenergic receptors. Dosage is 6 mg for adults and 3 mg for the elderly. The most common side effects are somnolence, headache, nausea, and occasionally upper respiratory tract infection. The effects of doxepin use on plasticity are discussed in the section concerning off-label insomnia drugs with the other antidepressants.
3.2.3 Melatonin Receptor Agonists
Over the counter melatonin formulations vary in purity and strength. Melatonin taken 30–120 min before sleep shortens latency to persistent sleep (LPS) but is not effective in maintaining sleep (Olde Rikkert and Rigaud 2001). Half-life in the immediate release formulations is 30–50 min versus 2 h in the sustained release formulations. It is used to treat acute or chronic circadian rhythm disturbances in normal or blind children. Reported doses for melatonin range from 1 mg in infants, 2.5–3 mg in older children, and 5 mg in adolescents: Doses ranging from 0.5 to 10 mg have been used in children with special needs (Owens 2009). In adults with delayed sleep phase syndrome, 0.5 mg taken 5–7 h before sleep onset may shorten sleep-onset delays. Phase shifting effects of melatonin are attributed to melatonin receptors MT1 and MT2, which are highly prevalent in the retina and suprachiasmatic nucleus. Melatonin impairs performance in some neurocognitive tasks such as reaction time, tracking, vigilance, and spatial memory.
Ramelteon, a synthetic melatonin receptor agonist (MT1 and MT2 receptors), is FDA-approved for the treatment of sleep-onset insomnia in adults, and for sleep maintenance insomnia only by the European Community Agency, but is not approved for use in children and adolescents. The recommended nightly dose for treatment of adults with insomnia is 8 mg. Safety data from animal models suggest that ramelteon has a low risk of cognitive impairment and physiological dependence (France et al. 2006; Rajaratnam et al. 2009a, b). Efficacy in pediatric autistic cases has been reported (Owens and Mindell 2011). Peak absorption is within 0.75–1 h, and half-life ranges from 0.8–2.5 h (Karim et al. 2006). Main side effects include somnolence, dizziness, and fatigue. Ramelteon does not produce tolerance, withdrawal, rebound insomnia, cognitive impairment, or psychomotor retardation. It can be used in patients with substance abuse histories as well as in patients with mild to moderate pulmonary disease. It is metabolized by cytochrome (CYP)1A2 and should not be used with fluvoxamine. It does not significantly affect memory, digit symbol substitution, dual attention, auditory tracking, letter cancellation, or driving performance (Mets et al. 2011).
Non-24 h sleep–wake disorder (hypernyctohemeral syndrome) is a circadian rhythm sleep disorder that affects individuals who lack the capacity to synchronize their circadian clock with the external environment. Total blindness, major psychiatric disorders, and structural lesions of the hypothalamus are comorbid conditions. Affected individuals present with difficulty in initiating sleep and progressively delayed sleep onset and offset. Tasimelteon is the only drug that is currently approved by the FDA to treat non-24-h sleep–wake disorder; the recommended dose is 20 mg at night before bedtime, taken without food. Tasimelteon is a MT1/MT2 agonist with a higher affinity for the MT-2 receptor; its elimination half-life is 1.3–3.7 h, and peak concentration is at 2 h; it is metabolized mainly by CYP1A2 and CYP3A4 enzymes (Bonacci et al. 2014). Tasimelteon (10, 20, 50, 100 mg) reduced sleep latency and increased sleep efficiency in healthy adults and in adults with transient insomnia; WASO significantly improved at higher doses (20, 50 mg). Phase shifting was dose dependent. Adverse events included somnolence and headache, but were similar to placebo (Rajaratnam et al. 2009a, b). Twenty milligram of tasimelteon at night entrained the master clock, reduced daytime sleepiness, increased nighttime sleep, and optimized sleep timing in 84 blind participants with non-24 h sleep disorder (Johnsa and Neville 2014). Tasimelteon was also better than placebo in maintaining entrainment and continuing to improve sleep and wake parameters (Johnsa and Neville 2014).
Agomelatine, a melatonergic MT1/MT2 agonist and an antagonist for 5-HT2B and 5-HT2C, improves insomnia associated with depression in manic-depressive (MD) disorders and major depressive disorders (MDD). By blocking 5-HT2C receptors, agomelatine induces release of both NE and DA. At a dose of 25 mg/day, agomelatine improved sleep efficiency, slow-wave sleep, and distribution of delta activity throughout the night, but did not affect REM sleep latency or amount of REM sleep (Quera-Salva et al. 2010). Agomelatine has been approved in Europe for several years, but development for the US market was discontinued after the results of Phase III trials. Agomelatine appears similar in efficacy to other antidepressants but requires liver monitoring because of 1.3 % incidence of elevated liver enzymes and 6 cases of non-fatal toxic hepatitis had been reported (Taylor 2014).
Importantly, there is evidence from experimental studies in animals indicating that melatonin and melatonin analogs may affect neuronal plasticity. In rats, subjected to experimental stroke or glutamate excitotoxicity, melatonin administration improved neuroplasticity in cultured neurons by significantly upregulating growth associated protein-43, NMDAR postsynaptic density-95, and matrix metalloproteinase-9 protein (Juan et al. 2013). Also, chronic agomelatine administration enhanced hippocampal cell proliferation and survival in stressed rats but not in control rats (Dagyte et al. 2010). On the other hand, in other studies, ramelteon did not affect NREM or REM sleep, did not affect ocular dominance plasticity, and did not affect learning or memory (Seibt et al. 2008; Hirai et al. 2005).
3.2.4 Off-label Insomnia Drugs
Of the four most commonly prescribed hypnotic agents, three are antidepressants that are prescribed off-label: trazodone, amitriptyline, and mirtazapine (Walsh 2004). Tricylic antidepressants (TCAs) such as amitriptyline and doxepin (1) inhibit serotonin (5 HT) and norepinephrine (NE) reuptake transporters; (2) desensitize presynaptic 5 HT1A autoreceptors, sensitize postsynaptic 5 HT1A receptors, and both downregulate and antagonize postsynaptic 5 HT2 receptors; (3) desensitize presynaptic α2 autoreceptors and downregulate postsynaptic β receptors; (4) antagonize peripheral α1 and α2 adrenergic receptors. The net effect of TCAs is to enhance serotonin and norepinephrine neurotransmission in the central nervous system (Buysse 2011). Sedating TCAs also antagonize H1 histamine receptors and muscarinic M1 cholinergic receptors.
Antidepressant drugs, which are often used to treat insomnia, are known to affect neuronal plasticity. Antidepressant drugs reverse the structural and functional consequences of stress in the hippocampus and prefrontal cortex, but do not reverse the changes observed in the amygdala (Pilar-Cuéllar et al. 2013). Also, the antidepressant efficacy has been linked to increased levels of brain-derived neurotrophic factors, which in turn may promote hippocampal neurogenesis (Castren and Hen 2013; Lucassen et al. 2010; Wainwright and Galea 2013). Chronic antidepressant therapy induces increased precursor proliferation in the dentate gyrus and increases survival of newborn neurons that functionally integrate into hippocampal circuitry. Synaptogenesis and synaptic elimination are promoted through arborization and pruning of axonal and dendritic branches. Antidepressants also influence plastic regulation for synaptic strength. Information transfers through the active synapses via LTP, while inactive synapses are suppressed via long-term depression. Environmental activity regulates transcription and translation of effector genes involved in neuronal plasticity (Castren and Hen 2013).
Also, cognitive function can be affected by antidepressant medications. In remitted major depressive disorder subjects, a recent study evaluated memory and executive functions in 4 groups of subjects: 50 medicated (29 treated with tricyclic antidepressants (TCAs), 21 with selective serotonin reuptake inhibitors (SSRIs) or serotonin noradrenaline reuptake inhibitors), 19 medication-free subjects, and 31 controls (Nagane et al. 2014). All 3 depressed groups had significantly lower performance for verbal memory compared with controls. Visual memory was worse in both medicated groups compared to controls, while no significant difference was found between controls and the non-medicated group. Only the group treated with TCAs showed a significantly lower performance in cognition compared with controls.
Trazodone, a tetracyclic antidepressant, weakly inhibits serotonin reuptake transporters, but has no effect on dopamine or NE reuptake. Trazodone inhibits serotonin receptors (5-HT1A, 5-HT2A, and 5-HT2C), adrenergic receptors (α1 and α2), and histamine receptors (H1). It has sedative properties and improves subjective sleep quality. Effects on sleep latency, TST, and sleep efficiency are inconsistent; slow-wave sleep is increased; REM sleep is not significantly changed (Buysse 2011). In cats, trazodone significantly impaired sleep-dependent consolidation of ocular dominance plasticity: Deficits occurred in both the normal depression of V1 (medial occipital lobe) neuronal responses to deprived eye stimulation and potentiation of responses to non-deprived eye stimulation (Aton et al. 2009). These results were hypothesized to be due to trazodone blockade of 5-HT2C receptors during sleep, which impairs the normal strengthening and weakening of synapses in V1 (Aton et al. 2009).
Mirtazapine is a presynaptic α2 adrenergic reuptake inhibitor; it increases noradrenergic tone through blockade of α2 adrenergic autoreceptors and heteroreceptors. Enhanced serotonergic neurotransmission is mediated via 5-HT1 receptors. Mirtazapine is a postsynaptic serotonergic 5-HT2 and 5 HT3 antagonists. Mirtazapine has weak muscarinic, anticholinergic, and histaminic (H1) antagonist effects. It decreases sleep latency, increases TST, and reduces WASO, in adults with and without depression. Mirtazapine is associated with sedation, weight gain, and dry mouth. With acute dosing at 30 mg, mirtazapine prolonged next-day motor reaction and impaired driving performance compared to placebo; sub-chronic administration did not affect performance (Wingen et al. 2005). Similar results were observed in another study comparing effects of mirtazapine 15 mg to trazodone 25 mg: Acute treatment with mirtazapine increased sleepiness and impaired road-tracking performance, but not car-following performance, while trazodone did not impair driving or cognitive performance (Sasada et al. 2013). Mirtazapine enhanced cognitive functions associated with prefrontal cortex in patients with recurrent depression, with 2/3 showing normal values after 6 months of treatment. This improvement did not correlate with the degree of amelioration of depression (Borkowska et al. 2007). Adjunctive mirtazapine added to first generation antipsychotics in a group of schizophrenic subjects improved cognitive function (Stenberg et al. 2010).
Amitriptyline is a potent REM sleep suppressant. Anticholinergic side effects can lead to blurred vision, dryness of the mouth, urinary retention, and orthostatic hypotension. Rapid withdrawal of TCAs can lead to REM rebound and nightmares. Amitriptyline overdose can be fatal within 24 h of ingestion due to cardiovascular toxicity with ECG abnormalities, arrhythmias, and hypotension (Thanacoody and Thomas 2005).
Gabapentin and pregabalin have analgesic and sedative effects; both have been associated with improvement in self-reported sleep measures in patients with painful conditions, such as fibromyalgia, neuropathy, post-surgery, and post-herpetic neuralgia (Buysse 2011). Both drugs do not interact with GABAA or GABAB receptors, but may promote GABA production in the CNS. Daily doses of gabapentin range from 300–2,100 mg, while pregabalin doses range from 150–600 mg daily; both are administered in divided doses, with larger doses at night. Side effects include sedation, fatigue, dizziness, ataxia, and less commonly leukopenia.
Tiagabine, an adjuvant anticonvulsant, inhibits GABA transporter GAT1 and increases the inhibitory actions of GABA (Buysse 2011). Doses of 4–16 mg have been utilized to treat insomnia. Dose-related increase in slow-wave sleep has been reported; effects on TST and WASO are inconsistent. Side effects include dizziness, nausea, and somnolence. A study comparing the effects of anti-epileptic drugs on associative LTP-like plasticity in the human motor cortex reported that gabapentin administration has no effect on LTP-like motor cortical plasticity, while tiagabine use resulted in non-significant trends toward reduction of long-term potentiation-like motor cortical plasticity (Heidegger et al. 2010).
Gaboxadol is a novel selective extrasynaptic agonist of GABAA receptors that contain α4, α6, and delta subunits. Gaboxadol has peak levels within 30 min of intake and has a half-life of 1.5–2 h. Doses of 5–15 mg reduced LPS and WASO, increased TST, and increased slow-wave sleep duration without significant effects on duration of Stage 1, Stage 2, or REM sleep (Deacon et al. 2007). Cognitive testing the next day after gaboxadol administration did not show any significant impairment in the cognitive drug research test battery results (Deacon et al. 2007). Gaboxadol did not alter information processing, psychomotor performance, and memory measured 12–24 h after treatment (Boyle et al. 2009; Deacon et al. 2007). On the other hand, animal studies suggest that Gaboxadol may have cognitive effects that depend on the duration of the treatment. While acutely increasing δGABAA activity in mice may result in memory impairment, long-term treatment may improve memory (Whissell et al. 2013). Particularly, pre-treatment with gaboxadol for 7 days improved discrimination memory (Whissell et al. 2013). Such memory enhancing effects are associated with an increase in the generation of new neurons in the hippocampus, which are thought to contribute to spatial memory, recognition memory, and fear memory (Marín-Burgin and Schinder 2012).
Clonidine, a central α2 agonist, decreases adrenergic tone. It is commonly prescribed for sleep-onset delay in children and for ADHD symptoms. Peak effects occur at 2–4 h and interrupted sleep may occur as levels drop (Owens and Mindell 2011). If tolerance develops, dosage may be increased. Clonidine decreases REM and slow-wave sleep; discontinuation can precipitate REM rebound or slow-wave sleep rebound. Side effects include hypotension, bradycardia, anticholinergic effects, and dysphoria. This medication should be avoided in diabetic patients and patients with Raynaud’s syndrome.
Positron emission tomography measurements of brain activity in healthy subjects following clonidine infusion showed reduced functional strength of fronto-thalamic connections and pathways to and from the visual cortex at rest (Coull et al. 1999). While performing attentional tasks, clonidine increased the modulatory effects of frontal cortex on projections from locus coeruleus to parietal cortex (Coull et al. 1999). Clonidine improved memory and task performance in Korsakoff’s amnesia patients and schizophrenic patients (Mair and McEntee 1986; Fields et al. 1988). In schizophrenic patients, reduced pre-pulse inhibition of the startle reflex may be a manifestation of reduced filtering of sensory information, which can lead to cognitive fragmentation, hallucinations, and delusions. Clonidine improved and normalized these pre-pulse deficits (Oranje and Glenthoj 2013). Clonidine also facilitated mnemonic functions and spatial working memory in parkinsonian patients, although planning, single spatial span memory, or spatial recognition memory did not improve (Riekkinen and Riekkinen 1999).
Quetiapine is an atypical anti-psychotic that is commonly used to treat insomnia in patients with psychiatric illness and dementia and for insomnia in prisoners and military personnel (Anderson and Vande Griend 2014). It is an antagonist with strong affinity for H1 receptors, moderate affinity for 5-HT2A receptors and 5-HT1A receptors, weak affinity for adrenergic α1 and α2 receptors, dopamine receptors D1 and D2, and has no appreciable affinity for muscarinic, cholinergic, or benzodiazepine receptors. Its half-life is 2–3 h. Although antipsychotic dosages range from 150–800 mg daily, off-label use for insomnia ranges from 25–200 mg. Twenty-five mg of quetiapine reduced mean sleep latency and increased mean TST in 13 healthy men compared to placebo group; although a trend for improvement was demonstrated, statistical improvement was not demonstrated (Tassniyom et al. 2010). Quetiapine (25, 100 mg) improved subjective measures of sleep quality, sleep latency, and sleep continuity in 14 healthy men. Polysomnography showed increased TST, increased sleep efficiency, and increased percentage for Stage 2 sleep; periodic leg movements of sleep were significantly increased with the 100 mg dose (Cohrs et al. 2004). Side effects include dry mouth, weight gain, and increased blood sugar.
Neuronal and synaptic plasticity may be altered with quetiapine. Hippocampal neuronal cultures in rats showed that atypical antipsychotics (including quetiapine) up-regulated synaptic proteins and dendritic outgrowth (Park et al. 2013). fMRI studies in schizophrenics before and after 12 weeks of quetiapine monotherapy showed that treated patients had a significant increase of activation in the ventrolateral prefrontal cortex, lingual gyrus, and right precuneus. Behavioral measurement of responses including reaction time and performance demonstrated slight improvements, but these did not reach statistical significance (Meisenzahl et al. 2006).
3.2.5 Unregulated Agents for Insomnia
Alcohol is used as a sleep-aid by 6–13 % of the US population. Unregulated health supplements used to improve sleep include valerian root (Valeriana officinalis), kava-kava (Piper methysticum, lavender, hops (Humulus lupulus), passionflower (genus Passiflora), lemon balm (Melissa officinalis), and skullcap (genus Scutellaria) (Weeks 2009). Extracts of magnolia and phellodendron bark have mild sedative effect. Supplements, such as GABA, theanine, tryptophan, and 5 hydroxytryptophan are purported to promote relaxation (Weeks 2009). As these supplements are unregulated, purity and concentration may vary across formulations. A 4-way crossover study in 9 healthy subjects compared valerian (500, 1,000 mg) to triazolam 0.25 mg and placebo. Interestingly, valerian did not affect either cognitive or psychomotor performance at the doses used, while triazolam had detrimental effects on cognitive processes (Hallam et al. 2003). A review of neurocognitive effects of kava evaluated 10 studies (7 acute, 3 chronic use) and concluded that majority of the studies did not demonstrate replicable significant negative effects on cognition (LaPorte et al. 2011). One acute study found that kava improved visual attention and working memory processes, while another showed that body sway was increased. One chronic study found that kava significantly impaired visual attention during high cognitive demand (LaPorte et al. 2011).
3.2.6 Hypocretin (Orexin) Antagonists
In recent years, the hypocretin/orexin system has received a great deal of attention as a target for insomnia treatment. Hypocretin-1 and hypocretin-2 (also known as orexin-A andorexin-B) are neuropeptides produced in the lateral hypothalamus. Hypocretin neurons receive input from nuclei mediating stress, autonomic tone, reward and motivation, as well as the circadian clock responsible for the timing of sleep and wakefulness. The hypocretin cells integrate these various inputs to promote many aspects of arousal via two different receptors (HcrtR1 and HcrtR2) (Scammell and Winrow 2011). Hypocretin neuronal activity oscillates during the active phase but is low during the normal sleep period. Hypocretin antagonists promote the transition between arousal and sleep by inhibiting the orexin-mediated wakefulness system that is responsible for the transition between arousal and sleep. Single hypocretin receptor antagonists have been studied (SB-334867 binds to HcrtR1 and JNJ-10397409 binds to HcrtR2), but only the dual hypocretin receptor antagonists (DORA) are undergoing FDA phase trials (Winrow and Renger 2014).
On August 13, 2014, the US Food and Drug Administration approved suvorexant as the first drug in its class to treat insomnia. Suvorexant, a potent HcrtR1-HcrtR2 antagonist, was approved at 4 different strengths (5, 10, 15, 20 mg) to treat sleep-onset and sleep-maintenance insomnia. Median peak plasma levels occur 2 h after ingestion; it is extensively metabolized primarily by CYP3A4 with some contribution from CYP2C19. The mean terminal half-life is about 12 h, while the drug’s accumulation under the curve (AUC) and maximum concentration (Cmax) is about 1.2–1.6 h at steady state. Oral clearance is about 20 % lower in women than in men. The concentration of suvorexant 9 h after dosing is 15 % higher in the elderly compared to the non-elderly and is 20 % higher in obese patients than in those with normal BMI (FDA 2013).
Three randomized, double-blind, placebo-controlled studies evaluated suvorexant at varying doses and with different study designs as a treatment for insomnia (Sun et al. 2013; Herring et al. 2012; Michelson et al. 2014). Suvorexant improved sleep maintenance with reduced WASO, increased sleep efficiency and TST, and improved sleep latency (Sun et al. 2013; Herring et al. 2012; Michelson et al. 2014). No statistically significant difference was found on EEG frequency bands, including delta activity based on power spectral analysis (Sun et al. 2013). All doses were superior to placebo, based on TST. All doses (10–80 mg) were superior to placebo in reducing WASO, but there was a dose response related to reducing sleep latency. Higher doses (40, 80 mg) were more consistently effective, while the 10 mg dose was the least effective across measures (Herring et al. 2012). Based on the analysis of combined data from 3 studies submitted including concentration analyses results, the FDA officials concluded that there was no dose or concentration response based on objective sleep latency values; they opined that there was little evidence to suggest that higher doses were substantially superior to 10–15 mg dose (FDA 2013).
Suvorexant was generally well tolerated, but higher doses (40, 80 mg) showed higher rates of side effects compared to lower doses (10, 20 mg) (Michelson et al. 2014). Side effects reported included somnolence, headache, dizziness, abnormal dreams, upper respiratory or urinary tract infection, and increased alanine aminotransferase. Somnolence was present in 13 % of suvorexant group [Michelson et al. 2014) and was more frequent at higher doses [40, 50, 80, 100] mg (Sun et al. 2013; Michelson et al. 2014)]. Higher doses (40 or 80 mg) were associated with sleep paralysis (n = 2) and visual hallucinations (n = 2) (Michelson et al. 2014). In the above 3 studies, there were no reports of cataplexy or somnambulism side effects.
The FDA-approved lower doses of suvorexant than were utilized initially in clinical trials due to safety concerns regarding next morning sedation, driving performance, and suicidal ideation (FDA 2013). High-dose suvorexant had an eightfold increase in somnolence compared to low doses; somnolence incidence rose for at least 60 days from start of therapy. A formal driving study showed next-morning impaired performance at doses of 20 and 40 mg with increased standard deviation of lane position (defined as SDLP ≥2.4 cm ~ the equivalent of a blood alcohol level of 0.05 %) (FDA 2013). Four women discontinued testing due to somnolence at 40 mg. Suicidal ideation/behavior was noted in 8/1,268 patients compared to 0/1,012 placebo subjects. Although an adjudicating committee (Michelson et al. 2014) ruled on 45 reported cases, no cataplexy was identified: The FDA consultant believed there was at least 1 subject who had probable cataplexy (on 40 mg of suvorexant, laughing triggered leg weakness lasting 5–30 s and was associated with somnolence).
Several new hypocretin antagonist drugs are still in development. GSK’s HcrtR1-HcrtR2 antagonist SB-649868 has undergone 6 Phase I trials and has completed 1 Phase II trial to treat insomnia (Bettica et al. 2012a, b, c). Two phase I dose escalation trials in 103 men (10–80 mg single dose, 5–30 mg repeat doses) reported an estimated half-life of 3–6 h, dose-dependent CYP34A4 inhibition (mild at 5 mg, strong at 30 mg), and somnolence/fatigue at higher doses (60, 80 mg) (Bettica et al. 2012a, b, c). Next-morning test was negative for residual cognitive effects, but fatigue was observed (60, 80 mg) (Bettica et al. 2012a, b, c). In a Phase II study, when compared to placebo, all doses of SB-649868 (10, 30, 60 mg) administered 90 min before bedtime significantly reduced sleep latency, but only higher doses (30, 60 mg) improved sleep maintenance (WASO). SB-649868 increased TST from 22–70 min, slightly reduced percentage of Stage I and slow-wave sleep, and dose-dependently increased REM sleep percentage (Bettica et al. 2012a, b, c). Most common adverse events included headache, dry mouth (dose-dependent), nasopharyngitis, and dose-dependent fatigue. There were no reported symptoms of hallucinations, sleep paralysis, or cataplexy. Next-day residual effects as assessed by cognitive tests and auditory verbal learning test gave mixed results except for reduction in the number of correct words at the end of the test (Bettica et al. 2012a, b, c).
There is some evidence from controlled studies in animals that hypocretin antagonists may affect neuronal plasticity and cognitive function. Compared to GABAA receptor agonists zolpidem and eszopiclone, a study in rat and rhesus monkeys concluded there is a wider therapeutic margin for sleep versus cognitive impairment using DORA. A wider therapeutic margin was shown by the expression of hippocampal activity-regulated cytoskeletal-associated protein, an immediate early gene product involved in synaptic plasticity (Uslaner et al. 2013).
Abuse potential is also a consideration in choosing a pharmacologic agent for treating chronic insomnia. Hypocretin signaling is an important element in the response to the reward circuits in the central nervous system that is activated by drugs of abuse. An experiment in rats showed that DORA prevented transcriptional and behavioral plasticity resulting from stimulant (amphetamine) exposure, particularly in the ventral tegmental area (Winrow et al. 2010, 2011) and also attenuated the capacity of nicotine to induce reinstatement of extinguished responding for a reinforce. These findings could be beneficial as it may prevent drug-induced plasticity and drug-relapse in substance abusers.
4 Narcolepsy and Its Pharmacological Therapy
Gelineau in 1880 first coined the term narcolepsy to describe a pathologic condition characterized by irresistible episodes of sleep of short duration recurring at close intervals, at times accompanied by falls triggered by strong emotion (Guilleminault and Abad 2009). Narcolepsy refers to a syndrome characterized by excessive daytime sleepiness, disturbed nocturnal sleep, and pathologic manifestations of REM sleep. These REM sleep abnormalities include sleep-onset REM periods, cataplexy, sleep paralysis, and hypnagogic hallucinations. Narcolepsy is newly categorized into narcolepsy type 1 (previously called narcolepsy with cataplexy) and narcolepsy type 2 (narcolepsy without cataplexy) to highlight the concept that the absence of hypocretin (orexin) is a fundamental marker of the disorder (American Academy of Sleep Medicine 2014).
4.1 Narcolepsy , Brain Plasticity , and Cognitive Function
The hypocretin system is critical in promoting wakefulness: Hypocretin deficiency is present in narcolepsy, and excessive somnolence is one of the hallmarks of this disorder. The neurons synthesizing hypocretin are affected by nutrition and behavioral states, and these factors lead to synaptic plasticity and reorganization of synaptic architecture in hypocretin neurons (Gao and Wang 2010). Results from TMS studies in narcolepsy patients showed a significant increase of relaxed threshold and active threshold, while central short period and central motor conduction time were unaffected; paired TMS showed a significant increase of short intracortical inhibition, and short intracortical facilitation was unchanged (Civardi et al. 2009). These changes in excitability of intrinsic circuits in motor cortex observed in narcolepsy patients could correlate with the deficit of hypocretin/orexin system, through a reduction of histaminergic hypothalamic inhibitory projections to cortical GABAergic neurons (Civardi et al. 2009).
TMS findings in drug-naive narcolepsy patients with or without cataplexy differ from those in treated patients. Resting motor threshold is a marker of the excitability of a central core of corticospinal motor neurons, excitatory interneurons, and target spinal motor neurons. A TMS study was performed on narcolepsy patients who were on central nervous stimulants (methylphenidate and/or modafinil) for EDS and either typical or atypical TCAs (clomipramine, fluoxetine) (Vijayakumari et al. 2013). Increased resting motor threshold was present in drug-naive narcolepsy patients compared to controls, suggesting lower motor cortex excitability and enhanced excitability of inhibitory circuits (Vijayakumari et al. 2013). With treatment, resting motor threshold became similar to that in controls. The change in resting motor threshold scores was greater in younger narcolepsy patients (Vijayakumari et al. 2013). The cortical silent period pre-treatment of narcolepsy was significantly higher compared to controls, suggesting reduced excitability of cortical networks in human narcolepsy. After treatment, the central silent period is decreased in narcoleptics but still significantly differed from the controls. It is hypothesized that the first part of central silent period may be due to spinal inhibitory mechanisms, while the second part may be of cortical origin (Vijayakumari et al. 2013). Another study using modafinil showed significant shortening of the central silent period with treatment, suggesting increase in motor excitability (Joo et al. 2010). Another parameter tested with TMS is central motor conduction time, which is an indirect measure of maximum conduction velocity of corticospinal axons. Central motor conduction time did not differ before and after treatment, nor was it different from controls (Vijayakumari et al. 2013).
A review of neuroimaging studies in narcolepsy with cataplexy cited the following results: (a) Hypothalamic anatomical and functional abnormalities are consistently found, compatible with loss of hypocretinergic neurons; (b) structural alterations are seen in the hypothalamus and limbic system particularly the amygdala and abnormal neural responses are noted in these areas during disruption of emotional processing associated with cataplexy; (c) involvement of the ventral tegmental area and nucleus accumbens in narcolepsy with cataplexy is demonstrated by both neurostructural changes and altered neural response during reward processing; (d) functional and structural changes in hippocampal and various cortical regions (particularly fronto-temporal) may relate to specific mood/cognitive disturbances seen in narcolepsy/cataplexy; and (e) reduced thalamic volume, perfusion, and glucose metabolism in narcolepsy/cataplexy might relate to diminished alertness and disrupted sleep architecture in these patients (Dang-Vu 2013).
4.2 Narcolepsy Treatment
Current therapy for narcolepsy is symptom-based. Scheduled naps can combat sleepiness, but are seldom sufficient as primary therapy.
4.2.1 Modafinil
Modafinil is standard therapy for hypersomnia with narcolepsy, with doses ranging from 200–600 mg daily. Armodafinil, its R-enantiomer, significantly improved daytime sleepiness in narcolepsy patients, as shown in short-term randomized trials as well as in a multicenter trial lasting >12 months (Black et al. 2010). Starting dose was 100 mg, and dose was titrated to a maximum dose of 250 mg daily.
Modafinil’s mechanism of action remains unclear, but is postulated to involve various neurotransmitter systems including dopamine, norepinephrine, histamine, glutamate, GABA, serotonin, and hypocretin/orexin. Modafinil increases sleep latency in the multiple sleep latency tests in narcoleptic patients and shift workers and also prolongs sleep latency in the maintenance of wakefulness test in narcoleptic patients and sleep apnea patients. Side effects include headache, insomnia, nausea, dry mouth, vomiting, tachycardia, palpitations, and rarely Steven–Johnson syndrome. Armodafinil has similar side effects, with headache in 25 %, nasopharyngitis in 17 %, insomnia in 14 %, and upper respiratory tract infection in 10 % of patients with excessive daytime sleepiness due to narcolepsy, sleep apnea, and shift work disorder (Black and Hirshkowitz 2005). Cardiovascular side effects were present in 5–9 % of all patients treated with armodafinil. Hypertension (6 %) and palpitations (3 %) were the most common adverse cardiovascular events reported (Black and Hirshkowitz 2005). Patients using modafinil or armodafinil who are on oral contraceptives are advised to use another form of contraception, while on modafinil and for 1 month after discontinuation, since modafinil can increase metabolism of oral contraceptives by inducing hepatic cytochrome P450 enzymes.
Modafinil improved information processing speed and increased energetic resources in the prefrontal region as demonstrated by low-resolution brain electromagnetic tomography (Saletu et al. 2009).
4.2.2 Amphetamines
Stimulants, such as amphetamine, methamphetamine, dextroamphetamine, and methylphenidate, are highly effective for sleepiness, but they are habit forming and have addictive potential.
At low doses, amphetamines increase catecholamine (dopamine and NE) release and inhibit dopamine and NE reuptake from presynaptic terminals. Presynaptic modulation of the dopaminergic system mediates EEG arousal effects. At higher doses, amphetamine increases release of serotonin and inhibits vesicular monoamine transporter. Use of these stimulants may result in tolerance and increased potential for addiction. Acute side effects include increase in systolic and diastolic blood pressure, anorexia, dryness of the mouth, palpitations and tachycardia, insomnia and restlessness, headaches, agitation, confusion, and dysphoria. Chronic use can lead to irritability and bad temper, profuse sweating, headaches, orofacial dyskinesia, chorea, and tremor. Methamphetamine can be neurotoxic at high doses.
Methylphenidate, an N-methyl derivative of amphetamine, has a shorter half-life and milder side effects. Side effects include psychosis, seizures, and anorexia; overdose can result in arrhythmias and sudden death.
The effects of prescription drug stimulants on plasticity remain under investigation. Amphetamine has a biphasic effect: Low doses enhance LTP in the prefrontal cortex, while high doses impair it (Xu et al. 2010). This may explain the improved cognition seen with low-dose amphetamine (0.25 mg/kg) administered to healthy subjects before performing a working memory task while undergoing fMRI scanning (Mattay et al. 2000) and improved reaction time on spatial working memory and Stroop tasks in schizophrenics and control subjects (Barch and Carter 2005). In contrast, chronic high-dose stimulant use (amphetamine, methylphenidate) is associated with impaired verbal learning and memory capacities (Reske et al. 2010). Repetitive use of amphetamine suppresses intrinsic neuronal excitability in the ventral subiculum, a part of the hippocampus that activates dopamine transmission; ventral subiculum synaptic output may be diminished, leading to impairment of ventral subiculum-dependent associative learning (Cooper et al. 2003). A review of 40 placebo-controlled studies of methylphenidate use in ADHD concluded that methylphenidate improved performance on planning/cognitive flexibility, attention/vigilance, and inhibitory control tasks in 71.4, 70.6, and 69.7 % of studies, respectively. Also, a total of 58.3 and 50 % of the studies that evaluated the effect of methylphenidate on tasks of memory and working memory/divided attention, respectively, noted improvement (Pietrzak et al. 2006).
4.2.3 Caffeine
Caffeine, an adenosine receptor antagonist, is the most common psychomotor stimulant used by the general population to improve alertness, mood, and cognitive performance. Sixty-three to eighty-two percent of narcolepsy patients use caffeine (Won et al. 2014). Caffeine tablets are available in 50–200 mg doses, and caffeinated beverages vary widely in content from 5–250 mg, with energy drinks containing as much as 505 mg. Peak levels occur within 0.5–1 h, and half-life is from 3–5 h. When taken before sleep, it delays sleep onset, decreases slow-wave sleep, decreases TST, and reduces sleep efficiency (Buysse 2011). High doses can lead to muscle twitches, cramps, and nervousness.
In rats, chronic low-dose caffeine treatment prevented short-term memory impairment and early LTP TP in acutely sleep-deprived rats (Alhaider et al. 2010). Low doses of caffeine (12.5, 50, 100 mg) enhanced task performance tests for simple reaction time and a rapid visual information processing task; all doses affected cognitive performance with a flat dose response effect for the above doses (Smit and Rogers 2000). In 12 healthy subjects, lower dose caffeine (250 mg) enhanced performance on the digit symbol substitution test and a tapping speed test compared to placebo, while high-dose caffeine (500 mg) produced less performance enhancement than the lower dose (Kaplan et al. 1997). Subjective effects were better (elation, peacefulness, pleasantness) with the 250 mg dose, while unpleasant effects (nervousness, anxiety, excitement, irritability, restlessness, palpitations, nausea) were more frequent in the 500 mg dose (Kaplan et al. 1997).
4.2.4 Histaminergic Drugs
Drugs which block histamine H3 autoreceptors (Pitolisant) increase brain histamine and enhance wakefulness in narcolepsy patients. The H3 receptors autoinhibit histaminergic neurons, thereby modulating the synthesis and release of histamine. As presynaptic heteroreceptors, H3 receptors also control the release of acetylcholine, glutamate, GABA, and peptides (Lin et al. 2011). Based on studies in KO mice, it is hypothesized that H3 R antagonists disinhibit H3 autoreceptors, thereby enhancing synaptic histamine, with activation of postsynaptic H1 receptors, thereby promoting wakefulness. In humans, administration of H3 receptor inverse agonist/antagonist pitolisant (BF2-649) increases histamine and noradrenaline neuronal activity and promotes wakefulness (Weisler et al. 2012; Schwartz 2011; Lazewska and Kiec-Konowicz 2010; Dauvilliers et al. 2013). Pitolisant (individualized dose 10, 20, 40 mg/day) improved somnolence in 26 adult narcolepsy patients and reduced Epworth sleepiness scale (ESS) scores by ~5 units; cataplexy frequency also decreased in those who continued to take it optionally for 9 months. Pitolisant has also been successfully used to treat refractory hypersomnia in a few cases of narcoleptic teenagers (Inocente et al. 2012).
A randomized study of narcolepsy patients compared ESS scores of 32 patients on pitolisant (10, 20, 40 mg) to 33 patients on modafinil (100, 200 mg) to 30 patients on placebo. Over 8 weeks, ESS scores declined by −3.4 ± 4.2 in placebo versus −5.8 ± 6.2 in pitolisant group, and −6.9 ± 6.2 in the modafinil group (Dauvilliers et al. 2013). Adverse events for pitolisant were headache (35 %), insomnia, abdominal pain, nausea, and diarrhea. For modafinil, adverse effects included abdominal pain, withdrawal such as symptoms, lymphadenopathy, and inner ear dysfunction (Dauvilliers et al. 2013). A Phase III trial NCT01789398 (Harmony IV) of narcolepsy patients with pitolisant added to oxybate is ongoing. Another trial NCT01800045 is recruiting subjects to evaluate frequency of cataplexy attacks with pitolisant use compared to placebo.
Pitolisant also reduced ESS by ~6 units in 12 obstructive sleep apnea (OSA) patients who received 40 mg daily for 3 days (Schwartz 2011). Pitolisant had a favorable risk/benefit ratio in 23–38 % of idiopathic hypersomnia and symptomatic hypersomnia patients who were refractory to the usual stimulants; however, mean ESS scores only decreased by 1.5 units in idiopathic hypersomnia patients (Leu-Semenescu et al. 2014).
Other H3 antagonists are still in development. APD916’s Phase I trial in 24 healthy volunteers was a randomized, double-blind, placebo-controlled study using doses of 1, 3, and 5 mg of APD916. APD916 had a half-life of approximately 50 h. Abnormal dreams, visual, and tactile hallucinations were reported at the 3 and 5 mg doses, and insomnia was common at the 1 mg dose.
GSK 189254, a potent H3 antagonist, increased wake, reduced slow-wave sleep, and decreased paradoxical sleep in rats. A safety and dose escalation multicenter trial NCT 00366080 trial of GSK 189254 was terminated but the reason for termination was not specified.
4.2.5 Antidepressants, Serotonergic Drugs, and Monoamine Oxidase Inhibitors
TCAs such as imipramine and SSRIs such as venlafaxine and reboxetine may be effective treatments for cataplexy, sleep paralysis, and hypnagogic hallucinations.
TCA’s inhibit reuptake of catecholamines, suppress REM sleep, and increase muscle tone. Abrupt withdrawal from TCAs can result in rebound, severe cataplexy.
SSRIs inhibit serotonin reuptake in the presynaptic cleft and also inhibit REM sleep. Noradrenergic reuptake inhibitors, such as venlafaxine, reboxetine, and duloxetine, have also been utilized to treat cataplexy, sleep paralysis, and hypnagogic hallucinations.
Ritanserin, a 5-HT2 receptor blocker, may be effective as an add-on drug at doses of 5–10 mg/day to combat sleepiness in narcolepsy. Ritanserin increases NREM slow-wave sleep. Selegiline, a monoamine oxidase B inhibitor, at doses of 20–40 mg may be effective treatment for cataplexy and daytime sleepiness, but has potential drug and diet interactions (Morgenthaler et al. 2007).
4.2.6 Sodium Oxybate
Sodium oxybate (SXB) is the sodium salt of gamma hydroxybutyrate (GHB) that is effective for the treatment of cataplexy, daytime sleepiness, and sleep disruption (Morgenthaler et al. 2006); it may be effective for the treatment of hypnagogic hallucinations and sleep paralysis.
GHB is a metabolite that is concentrated in the hypothalamus and basal ganglia. GHB is also a precursor of GABA and may act as a neurotransmitter or neuromodulator; it has high-affinity binding with GHB receptors in the hippocampus and cortex (Van Amsterdam et al. 2012). GHB inhibits glutaminergic neurotransmission in neocortical and hippocampal neurons. At low doses, GHB increases glutamate release; however, supraphysiological or pharmacological doses lead to GHB receptor saturation and desensitization with decreased glutamate release (Van Amsterdam et al. 2012). GHB has 3 types of interactions with the GABAergic system: as a precursor of GABA, as a promoter of GABA release, and as a low-affinity GABAB receptor agonist (Snead and Gibson 2005). GHB has a biphasic effect in rats: Low doses increase dopaminergic firing, while high doses inhibit dopaminergic cell firing (Diana et al. 1991). GHB is a weak but selective; at pharmacologic doses, GHB may interact with the endogenous opioid system and increase serotonin turnover. It increases slow-wave sleep, reduces arousals, and has a variable effect on REM latency and REM duration. At doses of 6–9 g SXB, cataplectic episodes decrease in frequency and daytime sleepiness improves. SXB requires split dosing at night. Common side effects include dizziness, headache, nausea, somnolence, confusion, and nocturnal enuresis.
In animal studies, GHB impaired learning and performance of memory tasks (Van Amsterdam et al. 2012). In rats, GHB administration had a biphasic effect: Low dose (10 mg/kg × 15 days) produced more neuronal loss in the prefrontal cortex and hippocampal CA1 regions compared to higher dose (100 mg/kg) (Pedraza et al. 2009). It is postulated that low doses of GHB increase release of glutamate with cytotoxicity on neighboring neurons, while higher doses initiate GABA mechanisms that inhibit the increased glutamate release, thereby exerting a protective effect on glutamatergic neurotoxicity (Mamelak 2007). Of concern also, is the age-specific effect of GHB (adolescent rats are more sensitive to GHB-induced cognitive effects), which are due to neuroadaptations in glutamatergic neurotransmission in the frontal cortex (Sircar et al. 2011). Animal studies, including proteomic and genomic studies, suggest that GHB increases oxidative stress, possibly inducing neurodegeneration (Van Amsterdam et al. 2012). Also, up- or downregulation of several proteins and genes may affect cognition, apoptosis, or neuroprotection (Van Amsterdam et al. 2012). A study in mice demonstrated accelerated functional recovery after focal cerebral ischemia by altering expression of several neuroplasticity related genes in the striatum (c-jun and neurocan genes) (Gao et al. 2008).
In humans, acute administration of GHB induced amnesia and impaired working memory and episodic memory in drug abusers and healthy volunteers (Carter et al. 2009). Heavy users of GHB report severe memory problems and case studies have reported short- and long-term memory impairment. However, the long-term effects of GHB on cognition in humans still need systematic investigation.
4.3 Treatment of Narcolepsy in Children
In a retrospective study of 51 children with narcolepsy who had been followed at Stanford Sleep Disorders Clinic from 2001 to 2009 or had participated in research with follow-up over a mean of 4.6 years, different medications including methylphenidate, protriptyline, amitriptyline, imipramine, clomipramine, fluoxetine, paroxetine, escitalopram, sertraline, atomoxetine), bupropion, and duloxetine were tried, but rarely continued. The most utilized and continued medications were modafinil (84 %), sodium oxybate (79 %), and venlafaxine (68 %) (Aran et al. 2010). In the past 4 years, atomoxetine, a noradrenergic reuptake blocker, has been frequently used in children for its alerting and anti-cataplectic effect. For children weighing less than 70 kg, start at 0.5 mg/kg/day and increase after minimum of 3 days, as needed, to a maximum of 1.2 mg/kg/day given in a single dose. For adolescents, start at 25 mg/day and increase after minimum of 3 days, as needed, to a maximum of the lesser of 1.4 mg/kg/day or 100 mg/day. Modafinil was effective in treating sleepiness but did not affect cataplexy. Sodium oxybate (off-label use) was effective for cataplexy and for sleep consolidation. Starting dose for SXB was 60–90 mg/kg/day, administered at night in split dosing (1/2 at bedtime and 1/2 administered 2.5–4 h later) and titrated every 1–2 weeks to efficacy or development of side effects; maximal dose was 180 mg/kg/day, not to exceed 9 g/day. Urinary incontinence was not uncommon at the start, prompting dose reduction and slower escalation of dosage. Almost 20–25 % of children treated with SXB lost weight, while those on venlafaxine or modafinil did not. Children with narcolepsy and SDB were treated with CPAP therapy, in addition to their narcolepsy treatment regimen.
5 Restless Legs Syndrome and Its Pharmacological Treatments
Restless legs syndrome (RLS) is a neurologic disorder present in 4–7 % of individuals characterized by all of the following: An urge to move the legs usually but not always accompanied by or felt to be caused by uncomfortable/unpleasant leg sensations; beginning or worsening of urge/sensations during periods of rest or inactivity; partial or total relief of urge/sensations by movement (at least as long as activity continues); occurrence or worsening of urge/sensations during evening or night than during the day; and urge/sensations are not solely accounted for as symptoms of another medical or behavioral condition (Allen et al. 2014). The pathophysiology of RLS remains under investigation but altered dopaminergic function and insufficient brain iron have been implicated. It has been proposed that low cellular iron through direct or indirect mechanisms alters one or more components of the dopaminergic synaptic dynamics (postsynaptic receptors, presynaptic receptors, synthesis, release, or uptake of dopamine) resulting in compensatory changes in the other components (Earley et al. 2014).
About 70–80 % of RLS sufferers have primary RLS; secondary RLS can be associated with iron deficiency, end-stage renal disease, pregnancy, diabetes, peripheral neuropathy, and rheumatoid arthritis (García-Borreguero et al. 2007). Symptom severity can be classified into the following: (1) mild/intermittent, (2) moderate, (3) moderately severe/severe, or (4) refractory/intractable (Hening et al. 2009).
5.1 RLS and Plasticity
TMS studies have explored primary RLS pathophysiology. Single TMS pulse may yield a functional assessment of corticospinal conduction: Central motor conduction time is a measure of the integrity of the corticospinal pathways; resting motor threshold provides information about a central core of neurons in the primary motor cortex; and the amplitude of the motor evoked potential is an aggregate measure of the excitation state of output cells in the motor cortex. Motor threshold is increased if a significant part of the corticospinal tract is damaged and is decreased if the corticospinal tract is hyperexcitable. The cortical silent period is a measure of the suppression of the corticospinal output at a cortical level (Lanza et al. 2014). Meanwhile, paired-stimuli TMS studies investigate the intracortical inhibitory and facilitatory mechanisms within the motor cortex (Lanza et al. 2014). Several studies demonstrated significantly reduced short intracortical inhibition, suggesting impairment of cortical inhibitory circuits (Quatrale et al. 2003; Tergau et al. 1999). Using paired associative stimulation protocol by TMS, idiopathic RLS without treatment showed impaired associative sensorimotor plasticity; testing 4 weeks after dopaminergic therapy showed restoration of PAS plasticity (Rizzo et al. 2009). A review of TMS studies (Stiasny-Kolster et al. 2003; Quatrale et al. 2003; Scalise et al. 2006, 2010; Nardone et al. 2006; Rizzo et al. 2009) concluded that the corticospinal tract in RLS patients is unaffected (Lanza et al. 2014). Most studies (Tergau et al. 1999; Entezari-Taher et al. 1999; Quatrale et al. 2003; Scalise et al. 2006, 2010; Nardone et al. 2006; Kutukcu et al. 2006; Gorsler and Liepert 2007; Rizzo et al. 2009, 2010) demonstrated that resting motor threshold is not impaired in RLS patients, except for a study which reported a trend for increased active motor threshold in the tibialis muscle (Stiasny-Kolster et al. 2003) and a study (Gündüz et al. 2012) where decreased active motor threshold from the 1st dorsal interosseous muscle was noted only at night (Lanza et al. 2014). Many studies in RLS patients reported markedly reduced central silent period (Civardi et al. 2004; Joo et al. 2010; Das et al. 2013) that seems to be stable across the day. Treatment with dopamine agonists (Stiasny-Kolster et al. 2003; Kutukcu et al. 2006; Gorsler and Liepert 2007; Lanza et al. 2014) reversed this shortening. Intracortical facilitation is markedly reduced in RLS patients (Tergau et al. 1999; Quatrale et al. 2003; Scalise et al. 2004, 2006, 2010; Nardone et al. 2006; Rizzo et al. 2010).
Related posts:
 Memory Consolidation in Healthy Aging and Mild Cognitive Impairment
Restricted or Disrupted Sleep as a Causal Factor in the Development of Depression
Memory Consolidation in Healthy Aging and Mild Cognitive Impairment
Restricted or Disrupted Sleep as a Causal Factor in the Development of Depression
 and Electrophysiological Correlates of Sleep and Sleep Homeostasis
and Electrophysiological Correlates of Sleep and Sleep Homeostasis
 Bird’s Eye View of Sleep-Dependent Memory Consolidation
Bird’s Eye View of Sleep-Dependent Memory Consolidation
 and Synaptic Homeostasis
and Synaptic Homeostasis
 Studies of Sleep and Memory in Humans
Studies of Sleep and Memory in Humans
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


