Chapter 37 Tumors of the Pineal Region
• Pineal region tumors account for about 1% of adult and 3% to 8% of all pediatric intracranial tumors. They are rare but surgically treatable tumors that require surgical expertise, experience, and multimodality therapy.
• The histological variety of pineal region tumors consists of several major categories: (a) germ cell tumors (germinomas), (b) nongerminomatous tumors (embryonal carcinoma, choriocarcinoma, and teratoma), (c) pineal parenchymal tumors (pineoblastoma, pineocytoma, and papillary tumor), (d) tumors of the supporting structures (meningioma, ependymoma, astrocytoma, mixed glioma, choroid plexus neoplasm), and (e) other tumor types (metastases, cysts, lymphoma, variable).
• Diagnostic workup includes a thorough clinical and neurological examination, magnetic resonance imaging (MRI), and serum markers. Serum markers when positive are best used to follow the tumor response to therapy. Diagnostic approaches to pineal region tumors include stereotactic biopsy, endoscopic biopsy, and open surgery. In many cases, symptoms are caused by compression of the neural structures or obstructive hydrocephalus. Concomitant hydrocephalus can be treated by an endoscopic third ventriculostomy with biopsy, ventricular diversion/shunting, or surgical removal of the tumor.
• Pineal region tumors can be removed with relatively low morbidity using a variety or combination of approaches. The most popular ones are a posterior fossa approach (infratentorial-supracerebellar), supratentorial approaches (occipital transtentorial), and posterior and anterior transcallosal approaches. Surgeons should be familiar with the anatomical structures in the pineal region and with the indications, limitations, risks, and benefits of each approach. Each tumor requires an individualized surgical and treatment approach.
• Multimodal treatments such as radiotherapy, chemotherapy, and radiosurgery in combination with open microsurgery or stereotactic or endoscopic surgery should be incorporated into any treatment paradigm based on the extent of resection and histological type of tumor.
History
The mystique of the pineal gland’s function and the challenge of operating in this region make it worthwhile to provide a brief overview of the relevant historical facts. The anatomists of ancient times knew about the pineal body and named it konareion because of its cone-shaped appearance. It was first described by Herophilus of Alexandria (325-280 BC). Like all his contemporaries, he believed the ventricles to be the “seat of spirits” and thought that the pineal body acted like a sphincter, regulating the flow of thoughts between the third and the fourth ventricles. Galen (129-201 AD) was most likely the first to hypothesize that the pineal body served as a gland. During Galen’s time, the structures of the pineal region were compared with the external genital organs of men. They named the pineal body the “penis,” the superior colliculi of the quadrigeminal plate “testes,” and the inferior colliculli of the quadrigeminal plate “nates,” simply based on their comparable appearance to these structures. Gibson, in 1763, would later recapitulate these ideas in his Epitome of Anatomy.
Medical history therefore has three hypotheses about the function of the pineal gland:
1. A mechanical function, serving as a sphincter between the third and fourth ventricles
2. A spiritual function, serving as a seat for the spirit and the soul
Anatomy
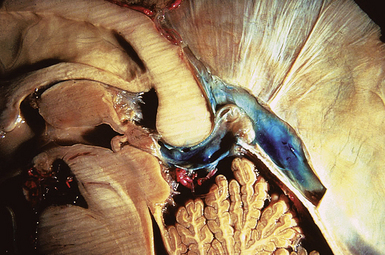
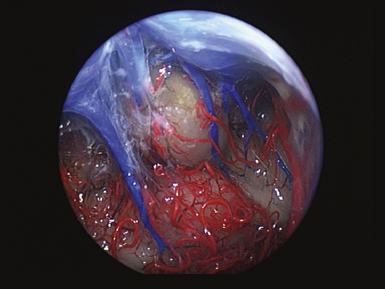
The pineal region is defined as the area of the brain bordered superiorly by the splenium of the corpus callosum and the tela choroidea, inferiorly by the quadrigeminal plate and midbrain tectum, anteriorly by the posterior parts of the third ventricle, and posteriorly by the cerebellar vermis as can be nicely shown in a midline sagittal section through the pineal gland and surrounding anatomical structures in an injected fixed human cadaver (Fig. 37.1). The pineal gland itself, as an invagination of the diencephalic ependymal roof between the habenular commissure and the posterior commissure, lies between the superior colliculi and basal to the splenium of the corpus callosum. It has a thin stalk to the epithalamus and developmentally is considered to be a paired structure. The neural connections with adjacent centers are the topic of discussion but have to be clarified. The pineal body itself has an average length of 7.97 mm (range 5.0-12.0 mm) and is 4.25 mm (2.5-7.0 mm) in height and width. It is oval in shape. The suprapineal recess projects posteriorly between the upper surface of the pineal gland and the lower layer of the tela choroidea in the roof. The stalk of the pineal body, from which the gland extends into the quadrigeminal cistern, has a cranial lamina and a caudal lamina. The habenular commissure, which connects the habenulae, crosses the midline over the cranial lamina, and the posterior commissure crosses the caudal lamina. The pineal recess projects posteriorly from the third ventricle into the pineal body between the two laminae. The posterior commissure forms the base of the triangular orifice of the aqueduct of Sylvius; the other two limbs are formed by the central gray matter of the midbrain. The quadrigeminal cistern is the subarachnoid space of the pineal region, and provides protection for the midbrain from the sharp edge of the tentorial notch. The arteries and veins inside the cistern are embedded in the firm arachnoidal septa. The quadrigeminal cistern communicates anteriorly with the cistern of the tela choroidea of the third ventricle, dorsally along the great vein of Galen with the dorsal cistern of the corpus callosum, laterally into the alae, caudally into the superior cistern of the cerebellum, which is formed by the medullary velum and the vermis, and anterodorsally into the ambient cistern. Through the latter cistern, the large arteries, the posterior cerebral artery and its branches, and the superior cerebellar artery approach the dorsal surface of the brainstem along the edge of the tentorium. The posterior border of the quadrigeminal cistern is formed by thickened opalescent arachnoid. Care must be taken when dissecting this area because the vein of Galen and its tributaries lie immediately rostral. The mesencephalon with the pineal body has its arterial blood supply from various sources. Small vessels supplying the parenchyma of the cerebral peduncles and the rest of the midbrain arise from the posterior communicating artery as perforating branches forming an internal or direct system. The vascularization in situ can be shown using neuroendoscopic techniques in injected human cadavers (arteries red, veins blue) and gives an impression of the enormous variety of the vascular structure (Fig. 37.2). All these arteries widely anastomose with each other, which explains the rarity of infarcts in the mesencephalon and the relative tolerance of the midbrain to direct surgical intervention. The pineal body is supplied by the pineal artery, which originates from the medial posterior choroidal artery, arising from the posterior cerebral artery (PCA), and enters the gland through its lateral portion from both sides. The great vein of Galen and its tributaries form a roof-like, dense venous network above the pineal body and the quadrigeminal plate, formed from the internal cerebral veins after they pass posteriorly through the tela choroidea and unite with the basal veins of Rosenthal. Other tributaries of the vein of Galen include the precentral cerebellar vein, internal occipital vein, posterior mesencephalic vein, and posterior ventricular vein. The straight sinus is formed by the joining of the great vein of Galen with the inferior sagittal sinus.
Histology
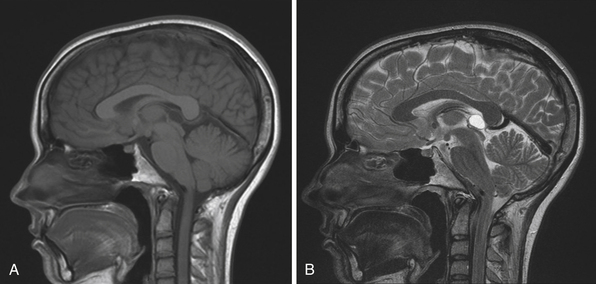
The normal pineal gland contains pinealocytes derived from amino precursor uptake and decarboxylation (APUD) cells and glial cells (astrocytes). The pineal gland is usually calcified by the age of 16 and appears so on routine computed tomography (CT) scans. Pineal cysts can be found incidentally in magnetic resonance imaging (MRI) studies in about 1.1% to 4.3% and this incidence seems to be higher in females (~2.4%) than in males (~1.5%) (Fig. 37.3). They can be identified in patients of all ages, with an increased prevalence found in older patients and in approximately 25% to 40% of autopsies, albeit many of the pineal cysts are microcystic. Their etiology remains obscure and may be the result of ischemic glial degeneration or of sequestration of the pineal diverticulum. Usually they are benign, non-neoplastic and may contain clear, xanthochromic, sometimes hemorrhagic, fluid. In the majority of cases, they are asymptomatic and may be followed on serial MRIs. Only in very rare cases do they show an enlargement, and may become symptomatic by causing hydrocephalus through aqueductal compression, headache, or gaze paresis, or hypothalamic symptoms. In those unusual situations with neurological symptoms or signs there is an indication to consider neurosurgical intervention.
Function
In reptiles, the pineal gland functions as a photoreceptor to change skin color in response to light. In humans, it is involved with hormone secretion for circadian rhythms and is known to have a neurotransmitter secretory function. The pineal gland inhibits gonadal development and regulates menstruation, adrenal function, and thyroid function. It is innervated by sympathetic nerves from the superior cervical ganglion that release norepinephrine to increase the pineal gland’s melatonin secretion. Light stimuli thereby reach the pineal gland from the retina via a polysynaptic pathway and affect the production of the neuroendocrine substance, melatonin. Melatonin, a derivative of serotonin, is essential in regulating circadian rhythms in endocrine gland activity and produces an antigonadal effect through the hypothalamic-hypophyseal axis. The production of melatonin is inhibited by light. Apparently, in many animals the major function of the pineal gland is to regulate the reproductive cycle; in fall and winter when days are short, more melatonin is produced, causing a gonad-suppressing effect. The exact function of the pineal gland in humans remains unclear. Recent studies could prove a diagnostic value of the melatonin profile in case of tumors of the pineal region. These studies showed a dramatically reduced melatonin rhythm in cases of undifferentiated or invasive tumors as well as the absence of melatonin variation as a consequence of pineal distortion after surgery. Also, the evidence for melatonin deficiency is recognized as a predictive factor to prevent post-pinealectomy syndrome. The absence of production of melatonin, as after pinealectomy, causes a “jet-lag-like” syndrome consisting of a complete disturbance of circadian rhythms.
Pathology
Pineal region tumors are rare tumors, with an estimated overall incidence of about 1% of all intracranial tumors (ranging from 0.5% to 1.6% in different studies) and are more common in children (3-8%) than in adults (less than 1%). Also, the incidence of pineal region tumors is higher among the Japanese, at 4-6% of all intracranial tumors. A population-based study calculated the incidence of pineal region tumors at 0.06 to 0.07 per 100,000 persons per year. No significant differences in the incidence between races were noted. Other reports describe the incidence in children younger than 20 years of age to be about 0.061 per 100,000 children per year. They are uncommon tumors and occur in a wide variety of different histological types. Furthermore, many tumors are of mixed cell type. The pathological classification depends on the substrate in the pineal region from which the tumor may arise. There are tumors which arise from the pineal glandular tissue itself, such as pinealocytomas (WHO grade I), pineal parenchymal tumors of intermediate differentiation (WHO grades II or III), pinealoblastomas (WHO grade IV), and papillary tumors of the pineal region. Other tumors arise from the glial cells, such as astrocytomas, oligodrendrogliomas, and glial cysts. Arachnoid cells are the basis for pineal-region meningiomas and non-neoplastic arachnoid cysts. Ependymal cells give rise to ependymomas in this region. Sympathetic nerves serve as a substrate for chemodectomas. Germ cell remnants give rise to germ cell tumors such as germinoma, choriocarcinoma, embryonal carcinoma, endodermal sinus tumor (yolk sac tumor), and teratoma. Lastly, the absence of a blood-brain barrier in the pineal gland makes it a suitable site for hematogenous metastases mostly from breast cancer, stomach cancer, renal cancer, or melanomas (Table 37.1).
TABLE 37.1 Types of Pineal Tumors: 2007 WHO Classification of Tumors of the Central Nervous System
| Tumor | Origin | Frequency |
|---|---|---|
| Germ Cell Tumors | Rest of germ cells | ~60% |
| Germinoma Mature Teratoma Immature Teratoma Teratoma with Malignant Transformation Yolk sac tumor (endodermal sinus tumor) Embryonal carcinoma Choriocarcinoma | ||
| Pineal Parenchymal Tumors | Pineal glandular tissue | ~30% |
| Pineocytoma (WHO grade I) Pineal parenchymal tumor of intermediate differentiation (WHO grade II or III) Pinealoblastoma (WHO grade IV) Papillary tumor of pineal region | ||
| Tumors of Supportive and Adjacent Structures | ~10% | |
| Astrocytoma Glioma (glioblastoma or oligodendroglioma) Medulloepithelioma | Glial cells | |
| Ependymoma Choroid plexus papilloma | Ependymal lining | |
| Meningioma | Arachnoid cells | |
| Hemangioma Hemangiopericytoma or blastoma Chemodectoma Craniopharyngioma | Vascular cells | |
| Non-neoplastic Tumor-like Conditions | <1% | |
| Arachnoid cyst | Arachnoid cells | |
| Degenerative cysts (pineal cysts) | Glial cells | |
| Cysticercosis | Parasites | |
| Arteriovenous malformations | Vascularization | |
| Cavernomas Aneurysms of the vein of Galen | ||
| Metastases | Absence of blood-brain barrier | <0.1% |
| Lung (most common), breast, stomach, kidney, melanoma | ||
Louis DN, Cavanee WK, Oghaki H. WHO Classification of Tumours of the Central Nervous System, 4th ed: WHO Classification of Tumours v. 1 (IARC WHO Classification of Tumours), World Health Org; 4th edition (May 2007).
Germ Cell Tumors
Germ cell tumors derive from pluripotential germ cells and span a wide range of differentiation and malignant characteristics. They constitute a unique class of rare tumors that affect mainly children and adolescents. Predominantly they occur in the midline when they arise in the central nervous system (CNS). Intracranial germ cell tumors, in general, account for 0.4% to 3.4% of intracranial neoplasms. Their average incidence is considered to be 0.1 per 100,000 persons per year. Pineal germ cell tumors occur primarily in males. Germ cell tumors in females are more common in the suprasellar region. Most germ cell tumors of the CNS occur in the first three decades (98%) with a peak in the second decade (65%) between 11 and 20 years. The histogenesis of germ cell tumors is a subject of controversy. For a long time, they have been assumed to represent the neoplastic offspring of primordial germ cells. During the last decade, however, a variety of speculative proposals have been published on this topic. As a result of the etiology, certain observations suggest that gonadotropins play a role in the development or progression of CNS germ cell tumors. These include the predilection with Klinefelter syndrome, a condition characterized by chronically elevated serum levels of gonadotropins. Due to the variety of possible origins, the following entities are distinguished: germinoma, embryonal carcinoma, yolk sac tumor (endodermal sinus tumor), choriocarcinoma, mature teratoma, immature teratoma, teratoma with malignant transformation, and mixed germ cell tumors. An accurate histological identification and subclassification of CNS germ cell tumors is critical for current treatment planning and prognostication. In fact, only the germinoma and teratoma are likely to be encountered as pure tumor types.
Germinoma
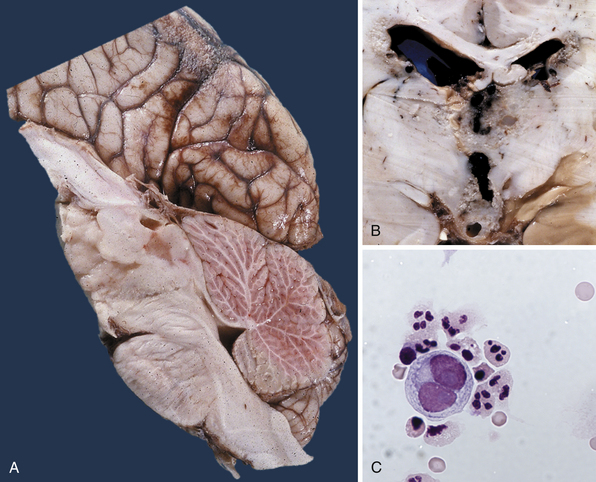
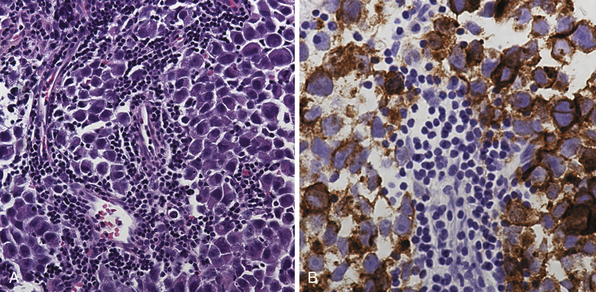
Pure germinomas (Fig. 37.4A to C) account for 65% to 72% of all intracranial germ cell tumors. These usually occur in the pineal region, although the second most common site is supra- and intrasellar, sometimes also parasellar. Germ cell tumors can be “synchronous,” and can be found both in the the suprasellar region and in the pineal region approximately 10% of the time. In fact, the discovery of a synchronous lesion on MRI is indicative of a germ cell tumor. The germ cell tumors are usually poorly circumscribed, light gray, granular, solid neoplasms that destroy the pineal gland and infiltrate the ventricular system and subarachnoid space early in their course. Hemorrhage into the tumor and necrosis or cystic degeneration are not often found. This tumor is composed of uniform cells resembling primitive germ cells, with large, vesicular nuclei, prominent nucleoli, and a clear, glycogen-rich cytoplasm. Additional features include lymphoid or lymphoplasma cellular infiltrates and, less frequently, scattered syncytiotrophoblastic giant cells (Fig. 37.5A and B).
Teratoma
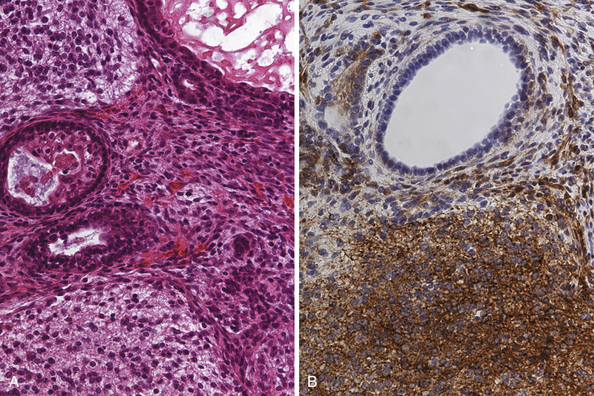
Teratomas (Fig. 37.6A and B) derive from all germ layers and are composed of well-differentiated tissues in an organ-like pattern. These tumors in the pineal region most often affect males. They are usually identified within the first two decades of life but occur more often in much younger children than other germ cell tumors (mostly in children younger than 9 years of age; about 20% occur between the ages of 16 and 18). By definition, the term teratoma can be used only when tumor elements derive from two or three germ layers. These tumors are usually well circumscribed, round or lobulated, and multicystic and compress the surrounding structures. The cystic component of the tumor may be watery, mucoid, or sebaceous. Sometimes bone, cartilage, hair, or teeth are present. Immature tumors are more frequently associated with a malignant course. Histologically, teratomas differentiate along ectodermal, endodermal, and mesodermal lines (e.g., they recapitulate somatic development from the three embryonic germ layers). Mature and immature variants as well as teratomas with malignant transformation must be distinguished from each other.
Immature Teratoma
This teratoma variant is composed of incompletely differentiated components resembling fetal tissue. Immature tumors are more frequently associated with a malignant course, but malignant potential may be explained by the presence of other germ cell tumor elements, such as choriocarcinoma or germinoma.
Pineal Parenchymal Tumors
Pinealocytoma (WHO Grade I)
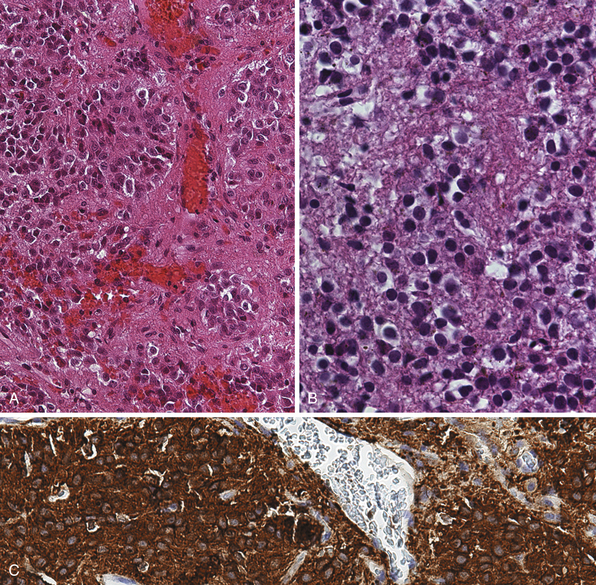
Pinealocytomas are defined as slow-growing pineal parenchymal neoplasms. They represent approximately 45% of all tumors with pineal parenchymal origin and occur throughout life, but most frequently in young adults (25-35 years). There is no sex predilection. Macroscopically, these tumors are well circumscribed with a gray-tan, homogeneous or granular cut surface. Degenerative changes, such as hemorrhage and small cystic cavities, can be present. They may disseminate widely along the CSF pathways. The histological appearance is of a well-differentiated neoplasm composed of small, uniform, mature cells resembling pinealocytes (Fig. 37.7A to C). They grow in sheets, but also feature large pinealocytomatous rosettes composed of abundant, delicate tumor cell processes. Pinealocytomas may contain astrocytic or neuronal components or both (so-called ganglioma of the pineal gland). Such tumors are the most benign of the pineal parenchymal neoplasms.
Pinealoblastoma (WHO Grade IV)
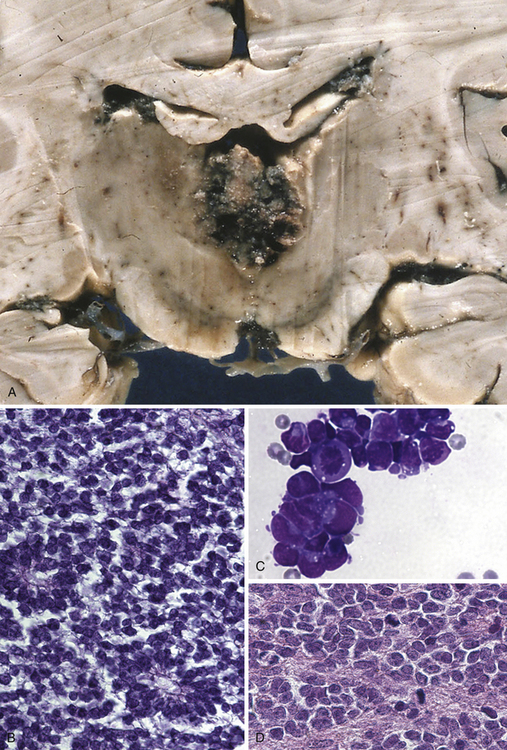
This type of tumor (Fig. 37.8A to D) is defined as a highly malignant, primitive embryonal tumor of the pineal gland itself and represents a true primitive neuroectodermal tumor (PNET). Although they are rare intracranial tumors, they constitute approximately 45% of all pineal parenchymal tumors. Usually they occur in the first two decades of life, more often in children, but principally can arise at any age. Some of these tumors may be present during the neonatal period. In larger series, pinealoblastomas are more common in males, with a male-female ratio of 2:1. The tumor usually replaces the tissue of the pineal gland. They are mostly soft, friable, and poorly demarcated and are pink, white, or gray, smooth or granular when cut, sometimes cystic, and frequently hemorrhagic or necrotic. Calcifications are rare and infiltration of the surrounding structures, including the meninges, is common. Constituting the most primitive of pineal parenchymal tumors, pinealoblastomas are composed of patternless sheets of densely packed small cells with round to irregular nuclei and scant cytoplasm. Pinealocytomatous rosettes are lacking, but Homer-Wright and Flexner-Wintersteiner rosettes may be seen. Retinoblastomatous differentiation of pinealoblastomas sometimes occurs and supports the theory of the photoreceptive origin of pineal gland cells. Pinealoblastomas may accompany bilateral retinoblastomas, in which case together they are called trilateral retinoblastoma. Pinealoblastomas sometimes contain melanin pigment. Because of its infiltrating nature into surrounding structures, dissemination in the CSF is not rare: according to the literature, between 8% and 24%. Metastases to bone, lung, and lymph nodes have been reported.
< div class='tao-gold-member'>






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


