Cell number
Amount of media for culture
Amount of media for a virus infection
1
6 well
1.1 × 106 in one well
2 mL
1 mL
2
T25
3 × 106
4 mL
3 mL
3
T75
1–2 × 107
14 mL
10 mL
4
T175
3–5 × 107
24 mL
20 mL
2.
At confluency, the passaged cells should have the documented quantity as listed. The number of cells may be quantified through a trypan blue exclusion method using a hemocytometer.
3.2 Propagating the Initial Culture from a Frozen Stock
1.
2.
Add 14 mL of growth media to a T75 flask.
3.
Thoroughly mix the growth media throughout the flask to ensure that it is evenly distributed.
4.
Thaw the frozen stock of HEK 293 cells in a water bath set to a temperature of 37 °C until it becomes completely thawed.
5.
Transfer the previously frozen, now thawed, cell line stock from the cryotube into the T75 flask containing the 14 mL of growth media.
6.
Incubate the HEK 293 cells with growth media in 37 °C, 5 % CO2, humidified incubator.
7.
After 20–24 h, aspirate and discard the growth media from the T75 flask. Immediately following, apply 14 mL of fresh and pre-warmed (in a water bath set to 37 °C) growth media. Please advise that it will take about 2–3 days for the HEK 293 cells to recover from the propagation and, thus, fully grow in a T75 flask.
3.3 Propagation (Subculture) of HEK 293 Cell Culture
1.
Aspirate and discard the existing growth media from the T75 flask and wash the attached cells with approximately 5 mL of PBS for the purpose of removing any excess of dead cells that were not removed through the initial growth media aspiration (see Note 4 ).
2.
Add 1.5 mL of 0.25 % trypsin-EDTA solution to the T75 flask containing the attached HEK 293 cells, thoroughly spreading it entirely across the flask, and incubate the flask until there is evident cell detachment present (placing the incubating flask into a 37 °C, 5 % CO2 incubator may accelerate cell detachment via trypsin activation).
3.
Add 5 mL of fresh 10 % FBS DMEM to the T75 flask, washing the cells on the flask’s side to the bottom of the flask.
4.
Homogenize the HEK 293 cell line culture in the growth media through repeated pipetting (the total volume of the T75 flask should be consistent with 6.5 mL).
5.
From one T75 flask, you can make 3× T75 flask (outlined in steps 6 and 7) or 1× T175 flask and 1× T75 flask (outlined in steps 8 and 9).
6.
From a single T75 flask, three separate T75 flasks can be processed. Aliquot the volume of the original T75 flask, which should be 6.5 mL in volume, to each of the three new flasks in an even manner.
7.
Add 12 mL of a fresh 10 % FBS, DMEM into each of the three T75 flask. The new, total volume should now be 14–15 mL.
8.
If using 1× T175 and 1× T75 add 5.5 mL from #4 into a T175 flask. Keep 1.5 mL in the existing T75 flask.
9.
Add 18 mL of growth media to T175 (total 24–25 mL) and 14 mL of media into T75 (total 14–15 mL) (of note, cells in the flask will grow a bit slower since the initial amount of cells will be lower).
10.
Spread the cells gently, yet thoroughly, over the bottom surface of the flask and return it to a 37 °C, 5 % CO2, incubator.
3.4 Generation of Frozen Stocks of HEK 293 Cells
1.
It is recommended to have at least 2.0–3.0 × 106 cells per 1 mL of freezing medium in a cryotube [4, 7]. As described in the Table 1, T75 flask can contain approximately 1.0 × 107 cells at confluency, ergo, three to four cryotubes of frozen HEK 293 cell stock can be processed. Please note that if the confluency is ill defined, it may be of particular interest to quantify the number of cells in order to achieve the desired 2.0–3.0 × 106 cells/mL [5, 8] (see Note 5 ).
3.5 Generation of a Shuttle Vector (Fig. 1)
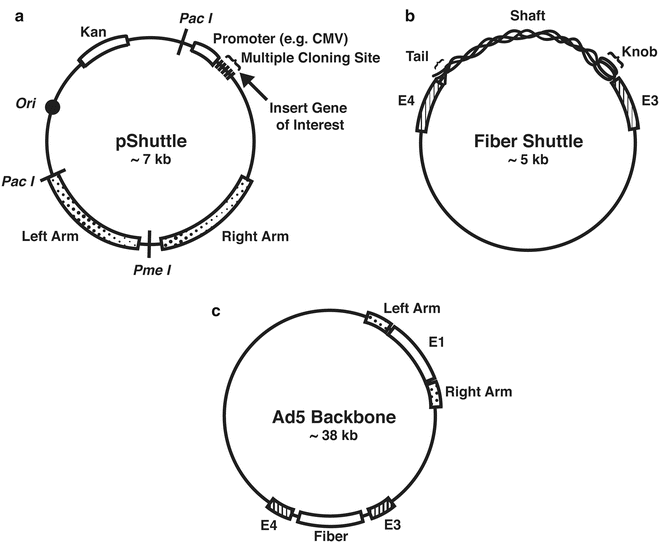
Fig. 1
Shuttle vectors and adenoviral backbone. (a) pShuttle vector(s) can be used to replace the E1 gene with the chosen gene of interest, thereby modifying the virus in a way that it is now replication incompetent. (b) A fiber shuttle vector can be used to modify the wild-type fiber, thus changing the viral tropism. (c) Commercial AdEasy or wild-type adenovirus type 5 may be used as a backbone
1.
Insertion of a gene of interest into the E1 region of the plasmid (see Notes 6 – 8 ). There are commercially available shuttle vectors that insert the gene of interest into E1 region. Through the usage of a multi-cloning site (MCS), a gene of interest can be inserted per the instructions of a standard molecular cloning method [3, 9–12].
2.
Modification of viral fiber(s) to alter viral tropism: Using the primers shown in Table 2 with adenoviral DNA as a template, the region of E3 (left arm)-fiber-E4 (right arm) can be obtained and inserted into any preferable common vector [3, 10–13].
Table 2
Primer set to generate fiber shuttle vector
Forward (SalI) | GCAGTCGACTCTAGAAATGGACGGAATTATTA |
Reverse (NotI) | ACGCGGCCGCACCGGGAGGTGGTGAATTACAA |
3.6 Generation of Adenoviral Vector with a Gene of Interest Through Homologous Recombination (Fig. 2)
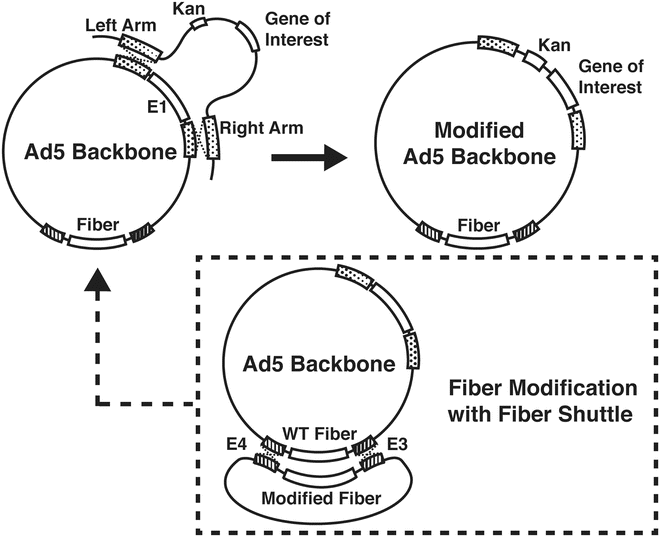
Fig. 2
Generation of plasmid DNAs of modified adenovirus by way of homologous recombination. Plasmid DNA of the adenoviral backbone and that of pShuttle (or pFiber shuttle) are co-transformed into BJ5183 cells. The resultants are selected by negative antibiotic resistance and the relative size of resultants’ DNAs (Fig. 3). Sequencing verified recombined DNA from BJ5183 cells should be retransformed into DH5α cells to achieve a large amount of stable DNAs
3.
Add PmeI-digested shuttle vector and Ad5 backbone (such as AdEasy): the ratio of DNA concentration is 3:1 in a maximum of 3 μl in volume.
4.
Transfer the mixture to the bottom of a pre-chilled, 1 mm gap electroporation cuvette.
5.
Electro-pulse the cuvette containing the mixture at 1800 V for 5 s.
6.
Resuspend the pulsed mixture in 1 mL of LB medium and transfer the mixture into 14 mL bacterial culture tube.
7.
Incubate at 37 °C for 40–50 min.
8.
Plate on 3 LB/Kan plates with 500 μl, 300 μl and 100 μl of the transformed mixture and incubate overnight at 37 °C (see Note 9 ).
9.
Pick 10–15 of the smallest, single (isolated) colonies and grow each in 2 mL of LB/Kan broth overnight at 37 °C with vigorous shaking (see Note 10 ).
11.
Transfer the bacteria grown LB into 1.5 mL tubes.
12.
Centrifuge at 14,000 rpm for 10 min.
13.
Aspirate all excess LB (a bacteria pellet should formed at the bottom).
14.
Add 300 μl of homogenizing buffer (commercially called P1 buffer) and homogenize well by thorough vortexing.
15.
Add 300 μl of alkaline buffer (commercially called P2 buffer) and mix through inverted shaking.
16.
Add 300 μl of acidic (neutralizing) buffer (commercially called P3 buffer) and mix through inverted shaking.
17.
Centrifuge at 14,000 rpm for 15 min.
18.
Transfer only the clear solution to a 1.5 mL tube (be cautious to remove any white debris). (However, if any white debris is collected, an additional centrifugation is necessary for its removal.)
19.
Add 560 μl of isopropanol and vortex.
20.
Centrifuge at 14,000 rpm for 15 min.
21.
Aspirate the supernatant (the DNA should be near the bottom of the tube).
22.
Add 400 μl of 70 % ethanol.
23.
Centrifuge at 14,000 rpm for 10 min.
24.
Aspirate out the supernatant (the DNA should be near the bottom of the tube).
25.
Dry completely.
26.
Add 20–30 μl of dH2O.
27.
Run on 1 % agarose gel (electrophoresis).
28.
Check the size of the plasmid DNAs (Fig. 3).
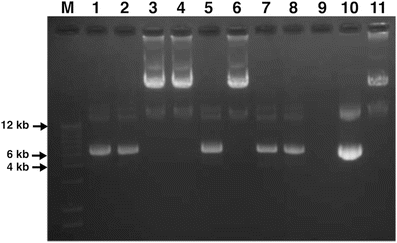
Fig. 3
Verification of homologous recombinants per gel electrophoresis. M: DNA ladder. 1–8: Homologous recombinants. 9: Void. 10: pShuttle vector. 11: Viral DNA of wild-type Ad5. After the homologous recombination of replacing the E1 gene region with the pShuttle vector (kanamycin resistant), plasmid DNAs of the resulting colonies were purified and separated on a 1 % agarose gel. Since the BJ cells can contain either homologous recombinants or the pShuttle vector, alone, and may grow in kanamycin LB, lanes 3, 4, and 6 are the possibly correct homologous recombinants, although verification by sequencing is required
29.
Verify via sequencing.
30.
Re-transform sequence verified DNAs into electro-competent DH5α or DH10B cells to obtain a large amount of stable DNAs (see Note 12 ).
3.7 Transfection of Adenoviral Vector in HEK 293 Cells
1.
Plate HEK 293 cells (or 911 cells) in a 6-well plate approximately 12–20 h prior to transfection. The confluency should be approximately 50–70 % at the time of transfection. Transfection of one well may be sufficient for virus generation and for further amplification. However, it is highly recommended that multiple wells be used for transfection as they will yield higher initial titers and quicker amplifications [7, 12, 17].
2.
 Lentivirus Production and Purification
Non-Viral, Lipid-Mediated DNA and mRNA Gene Therapy of the Central Nervous System (CNS): Chemical-Based Transfection
Gene Therapy for the Treatment of Neurological Disorders: Amyotrophic Lateral Sclerosis
Lentivirus Production and Purification
Non-Viral, Lipid-Mediated DNA and mRNA Gene Therapy of the Central Nervous System (CNS): Chemical-Based Transfection
Gene Therapy for the Treatment of Neurological Disorders: Amyotrophic Lateral Sclerosis
 Delivering Transgenic DNA Exceeding the Carrying Capacity of AAV Vectors
Delivering Transgenic DNA Exceeding the Carrying Capacity of AAV Vectors
 Gene Therapy of the Peripheral Nervous System: Celiac Ganglia
Gene Therapy of the Peripheral Nervous System: Celiac Ganglia
 Gene Therapy Models of Alzheimer’s Disease and Other Dementias
Gene Therapy Models of Alzheimer’s Disease and Other Dementias




Related posts:
Lentivirus Production and Purification
Non-Viral, Lipid-Mediated DNA and mRNA Gene Therapy of the Central Nervous System (CNS): Chemical-Based Transfection
Gene Therapy for the Treatment of Neurological Disorders: Amyotrophic Lateral Sclerosis
Delivering Transgenic DNA Exceeding the Carrying Capacity of AAV Vectors
Gene Therapy of the Peripheral Nervous System: Celiac Ganglia
Gene Therapy Models of Alzheimer’s Disease and Other Dementias
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
