Virology, Immunology, Transmission, and Disease Stage
Karl Goodkin
The Virus
Human immunodeficiency virus (HIV) is a retrovirus. Retroviruses have a ribonucleic acid (RNA) core. The term retrovirus refers to the fact that these viruses reverse transcribe their RNA to deoxyribonucleic acid (DNA) during the replication process. To accomplish this, HIV uses the enzyme reverse transcriptase (RT). Not coincidentally, this enzyme is a major target for anti-HIV (or antiretroviral) therapy. The predominant type of HIV worldwide is type 1 (HIV-1). HIV-1 has 9,749 nucleotides and is about the same size as other retroviruses. HIV-1 is further subdivided into subtypes (also known as clades), designated A through K (collectively referred to as group M), N, and O. More than 98% of HIV-1 infections in the United States are due to subtype B. Human immunodeficiency virus type 2 (HIV-2) has also been identified and likewise causes AIDS, though HIV-2 is not endemic in the United States. Originally, retroviruses were not thought to cause human disease. The HIV/AIDS epidemic has permanently changed that perception.
The HIV-1 genome has nine open reading frames, although 15 proteins are produced. The structural genes are gag, pol, and env. The gag gene and the gag-pol complex are translated into large polyproteins. These polyproteins are cleaved by a protease that is part of the Pol polyprotein. Gag polyprotein is cleaved into four proteins that are found in mature virions: matrix (MA), capsid (CA), nucleocapsid (NC), and p6 (a core protein). Pol polyprotein is cleaved into protease (PR) (a target for antiretroviral therapy), RT, and integrase (IN). The env gene codes for the envelope glycoprotein, gp160 (which comprises gp120 and gp41). The regulatory genes include tat, rev, nef, vpr, vif, and vpu. The gene tat (t rans-activator of tran-scription) is a positive regulator of viral protein synthesis and produces the protein Tat, which is known to have direct neurotoxic effects.1 The gene vpr (viral protein R) produces a protein that can bind glucocorticoid receptors, enhancing glucocorticoid resistance and increasing stimulation of the limbic-hypothalamic-pituitary-adrenal axis. vif (virion infectivity factor) is known to control viral infectivity; the protein Vif is needed for production of infectious virus because it inhibits an antiviral pathway in the cells that involves a host enzyme called APOBEC3G. The Rev (regulator of virion protein expression) and Nef (negative regulatory factor) proteins are involved in viral replication. Vpu (viral protein u), among other functions, enhances viral particle release from the host cell.
The glycoprotein coat of HIV-1, referred to as gp160 (molecular weight 160 kilodaltons [kD]) comprises two major proteins, the globular portion gp120 and the transmembrane portion gp41. gp120 is of great importance because it interacts with the primary target of HIV-1, the CD4+ T “helper” lymphocyte, a critical cell for immune system function. gp120 contributes to HIV-1 infection of the CD4 cell by binding to the CD4 receptor, which is followed by a conformational change and then by binding to a coreceptor on the surface of the cell (Figure 2.1). These coreceptors include cysteine-cysteine chemokine receptor 5 (CCR5) (on monocytes and macrophages) and cysteine-X-cysteine receptor 4 (CXCR4) (on lymphocytes). gp41 then changes its conformation, allowing membrane fusion to occur between the CD4 cell’s surface membrane and the viral glycoprotein envelope. It is through this mechanism that the viral core enters the cell. Once the core of HIV-1 has gained entry into the host cell, it is translocated to the nucleus. There, two outcomes may occur. The cell may begin a process of productive infection, or the cell may integrate HIV-1 into its genome (through the DNA complementary to the genomic RNA of HIV-1). In the latter case, the virus enters a latent (inactive) state (as a “provirus”).
Various factors may cause viral replication to be reactivated from the latent state. Latency is abrogated when the CD4 cell is stimulated during the normal immune response. One stimulant to reactivation of replication from the latent state is nuclear factor NF kappa B (NFKB), a host transcription factor. The latent state is known to be established in a number of cells and tissues (including CD4+ T helper lymphocytes in the peripheral blood and in macrophages in brain tissue). On reactivation from the latent state, viral messenger RNAs (mRNAs) are transcribed, viral proteins are translated, and whole virions are packaged. In the case of the CD4 cell, these virions bud off of the cell surface, resulting in a lytic form of cell death.
In addition to the glycoprotein envelope proteins described above, the virus also has core proteins, including p24, p17, p7, and p6. Of these, p24 (molecular weight 24 kD) is the best known; in an earlier stage of the epidemic this protein was quantified in serum to assess the probability of HIV-1 disease progression. The level of production of the core antigen
correlated well with the level of production of whole virions. Thus, it was used as a surrogate measure of viral replication, which is directly related to the likelihood of clinical disease progression.2 In fact, before the routine quantification of HIV-1 RNA copy number in plasma (“viral load”), the quantification of the p24 core antigen-enhanced by acid dissociation of antigen-antibody complexes (“complexed antigen”) before measurement-was felt to hold great promise as a measure of viral replication and proxy for clinical disease progression. This measure has remained useful and is currently recommended as a surrogate marker for plasma viral load for resource-limited settings internationally.
correlated well with the level of production of whole virions. Thus, it was used as a surrogate measure of viral replication, which is directly related to the likelihood of clinical disease progression.2 In fact, before the routine quantification of HIV-1 RNA copy number in plasma (“viral load”), the quantification of the p24 core antigen-enhanced by acid dissociation of antigen-antibody complexes (“complexed antigen”) before measurement-was felt to hold great promise as a measure of viral replication and proxy for clinical disease progression. This measure has remained useful and is currently recommended as a surrogate marker for plasma viral load for resource-limited settings internationally.
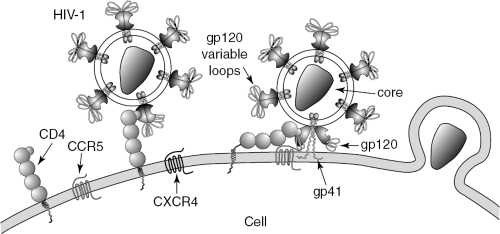 FIGURE 2.1 The hypervariable loop of gp120 (the globular portion of the envelope glycoprotein of HIV-1) binds to CD4 receptor (see interaction with HIV-1 at left). gp120 then changes its conformation and binds to one of the chemokine coreceptors for HIV-1 (e.g., CCR5 or CXCR4) (the latter in the interaction with HIV-1 at right). gp41 (the transmembrane portion of the envelope glycoprotein of HIV-1) then changes conformation, allowing fusion with the host cell membrane, culminating in viral entry (pictured at the far right). (Courtesy of HIV InSite, a project of the UCSF Center for HIV, hivinsite@ucsf.edu.) |
Process of Initial Infection
It was discovered early that HIV-1 infects cells expressing CD4 receptor on their surfaces, particularly T helper lymphocytes and monocytes in the blood. Monocytes differentiate into macrophages that express CD4 receptor on migration into the tissues. More recently, coreceptors required for the entry of HIV-1 into the cell were discovered. Of great importance are CCR5 and CXCR4. CCR5-using strains predominate in the early phase and infect monocytes, which are known to carry HIV-1 into the brain past an intact blood-brain barrier (mediating the subsequent infectious process in brain). CXCR4 is the coreceptor on the CD4+ T lymphocyte. CXCR4-using strains are associated with disease progression and predominate in later stages of HIV-1 disease.
When HIV-1 replicates in CD4 cells, the progeny virions produced bud off the cell, causing a lytic form of cell death. In macrophages, the cell is not killed; instead, it becomes a ware-house for a large number of virions. The presence of these virions affects the macrophage’s function in such a way that a chronic activated state is maintained. This activated state is associated with the secretion of several cytokines (soluble proteins released by immune cells that communicate with other cells). One of these cytokines, tumor necrosis factor-alpha (TNF-a) is associated with both demyelination and upregulation of HIV-1 replication. Another such cytokine, interleukin-1 (IL-1), is also associated with the upregulation of HIV-1 replication.
Systemic Viral Pathogenesis
HIV-1 depends on the CD4 cell for its replication and survival. As HIV-1 infection becomes established, antibodies are generated to its glycoprotein envelope. These antibodies are detected by the HIV antibody test, which is a combination of an enzyme-linked immunosorbent assay (ELISA) and a confirmatory, more specific test, the Western immunoblot assay. The HIV antibody test generally becomes positive an average of 6 weeks after exposure, although it can require as long as 6 months for this change to occur. Although longer periods for this process (termed seroconversion) have been described, this is now thought to be very rare.
When seroconversion has occurred and HIV-1 infection is initially established (versus an exposure after which no seroconversion occurs), the count of CD4 cells at this early point is generally maintained at a high level. However, this does not indicate a quiescent or latent stage, as was originally thought. Rather, the viral dynamics are very active at this stage, with 108 to 109 virions produced per day. The impact of this high level of viral replication is balanced by the capacity of the bone marrow to produce more CD4+ T lymphocytes at a daily rate of 2(10)9/day (5% of the body’s total). Although the CD4 cell count does not change significantly in the peripheral blood during this period, there is a loss of lymph node architecture due to a similarly high level of activity of HIV-1 infection in the lymph nodes. As the infectious process progresses, the ability of the virus to replicate itself eventually overcomes the ability of the bone marrow to replace the killed lymphocytes. Thereafter the CD4 cell count in the peripheral blood declines and immunologic disease progression is the result.
Systemic Immunopathogenesis
Early in the process of HIV-1 infection, the CD4 cell count remains in the normal range and there is no clinical manifestation of the infection. However, as viral replication with CD4+ T lymphocyte death overcomes the replacement of CD4+ T lymphocytes by the bone marrow, the CD4 cell count begins to decline in the peripheral blood. When the CD4 cell count falls below 500 cells/mm3, a clinically significant decrement has occurred. At this level of immune progression, the initiation of antiretroviral therapy has formally begun. However, more recently, the antiretroviral treatment guidelines have been modified to begin consideration of treatment at a CD4 cell count between 200 and 350 cells/mm3, depending on the level of plasma viral load.3 As the decline of CD4 cell number continues, the CD4 cell count eventually decreases below 200 cells/mm3, a landmark value. At this point, AIDS is defined on a laboratory basis, regardless of whether there has been any evidence of clinical disease progression.4 The risk for clinical disease is heightened at this severe immunologic stage of progression. Specifically, at this level, AIDS-indicator conditions commonly occur. Finally, at a CD4 cell count below 50 cells/mm3, a very severe level of immunoprogression has occurred in which the risk for mortality within 6 months increases.
The immunopathogenesis of HIV-1 infection is not solely related to the decline of the CD4 cell count over time. For example, the CD8+ T lymphocyte cell subset generally expands and becomes activated, as measured by coexpression of CD38 and HLA-DR.5 In addition, there is a shift in the pattern of cytokines produced by the CD4+ T lymphocytes over time. The level of cytokines produced by the subset known as Th1 cells decreases, while the level of those produced by the Th2 subset increases. As the Th1 cytokines (e.g., interferon-7 [IFN-7], IL-2, and IL-12) stimulate cellular immune functions, their decrement is associated with decreased immunologic monitoring of HIV-1 infection by cytotoxic T lymphocytes (CTLs) and systemic disease progression. The Th2 cytokines, in contrast, increase and are associated with stimulation of the humoral immune system and with abnormal, chronic, polyclonal B lymphocyte stimulation in HIV-1 infection. These changes are likewise associated with systemic disease progression.
Pathophysiology of Central Nervous System Disease
Before the introduction of today’s highly active antiretroviral therapy (HAART) regimens (now referred to as combination antiretroviral therapy [CART]), the cumulative prevalence of HIV-1-associated dementia (HAD) over the course of AIDS could be expected to be 21%, with an incidence rate during AIDS of about 7% a year.6 After the implementation of CART, the incidence rate of HAD has been documented to have decreased by 40% to 50% in the United States. The HAD prevalence rate has been suggested to have decreased to a lesser extent than the incidence rate because HAD patients are now living longer.
Minor cognitive motor disorder (MCMD) is a diagnosis that can properly be associated with the early symptomatic disease stage or with AIDS. It formerly was expected to be prevalent in as many as 25% of early symptomatic patients and as many as 50% of late symptomatic patients. However, recent data suggest that the incidence of MCMD, like that of HAD, has been reduced by CART. Currently, the cumulative prevalence rate of MCMD has been estimated at 14% in the early symptomatic stage and at 24.4% in AIDS.
HAD has been noted to be associated with ventricular enlargement and with cortical atrophy (as well as with brain atrophy generally). However, brain atrophy is not necessarily associated with the development of cognitive-motor disorder and is a nonspecific measure of risk for it. HIV-1 infection of brain has been widely described as a subcortical disease. In the basal ganglia, atrophy of the caudate nucleus has been noted to occur with HAD and has been specifically associated with decreased performance on neuropsychological (NP) tests requiring
psychomotor speed, manual dexterity, sustained attention, and flexibility of set shifting and sequencing. Likewise, white matter lesions are seen in HIV-1 infection of the brain. Although white matter hyperintensities on T2-weighted structural magnetic resonance imaging (MRI) imaging have been challenged regarding their clinical import, leukoariosis has been associated with decreased information processing speed. In contrast, cortical dysfunction has been thought to be restricted to late-stage HIV-1 disease in the brain.
psychomotor speed, manual dexterity, sustained attention, and flexibility of set shifting and sequencing. Likewise, white matter lesions are seen in HIV-1 infection of the brain. Although white matter hyperintensities on T2-weighted structural magnetic resonance imaging (MRI) imaging have been challenged regarding their clinical import, leukoariosis has been associated with decreased information processing speed. In contrast, cortical dysfunction has been thought to be restricted to late-stage HIV-1 disease in the brain.
HIV-1 penetrates into brain tissue as early as 2 weeks after infection, but rarely, if ever, directly infects neurons. Of seven brain regions examined in one important study, the hippocampus (Ammon’s horn) and the basal ganglia demonstrated the highest genomic HIV-1 RNA loads, with cerebellar and midfrontal cortices showing lower loads.7 An excess of multispliced RNAs over single-spliced RNAs suggests a contribution by abortive HIV-1 infection.8 Abortive infection in brain is related to the transient and restricted infection of astrocytes. Although abortive, this form of infection nevertheless expands the total viral burden in brain beyond the combination of genomic RNA (representing recent replication) and proviral DNA (representing the archive of virus infecting brain tissue).
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


