Voltage-Gated Ion Channels: Molecular Biology and Role of Mutations in Epilepsy
Dimitri M. Kullmann
Stephanie Schorge
Introduction
Voltage-gated ion channels have long been known to be central to the excitability of the brain. However, the recent discovery of several ion channel mutations in patients with seizure disorders (the “epileptic channelopathies”) has led to an appreciation of the importance of the molecular mechanisms underlying ion channel function in epilepsy. These experiments of nature have provided a unique insight not only into the mechanisms of seizure initiation and propagation but also into the fundamental molecular biology of ion channel synthesis and function.
What needs to be explained?
A parsimonious description of an epileptic channelopathy attempts to account for lowered seizure threshold on the basis of whether the mutation enhances or reduces the current carried by the channel (gain or loss of function, respectively). However, it is also important to explain the inheritance pattern associated with the mutation. Most of the known mutations associated with seizure disorders are either dominantly inherited or arise de novo in sporadic cases or in the parental germ line. This finding does not necessarily mean that recessive mutations do not occur: It could simply reflect the difficulty of identifying causative genes in kindreds with recessive inheritance.
Some seemingly monogenic epilepsies show extensive variability of clinical manifestations in different members of an affected family (Chapter 18). This variability is apparent as incomplete penetrance, or as phenotypic heterogeneity within a family carrying a particular mutation. Perhaps the best example of this phenomenon is seen in the genetics of a type of epilepsy associated with mutations in genes encoding sodium (Na+) channels. An important advance came with the demonstration that an extremely pleomorphic epilepsy syndrome, generalized epilepsy with febrile seizures (FS) plus (GEFS+), could be associated with a single mutation in a Na+ channel subunit109 (see the section Ion Channel Mutations in Epilepsy). GEFS+ is not a syndrome in the conventional sense because it cannot be diagnosed in an individual. Instead, it describes a spectrum of manifestations of abnormal central nervous system (CNS) excitability in different members of a family, ranging from uncomplicated FSs to malignant forms of epilepsy that can lead to profound mental and physical disabilities and premature death. At present, little is known about the relative importance of genetic and environmental factors in this variability. However, some recent efforts to identify modifying genes, summarized at the end of this chapter, provide a possible way forward.
Diversity of Voltage-Gated Ion Channels
Given the central role of ion channels in controlling neuronal excitability, and their roles as targets of several antiepileptic drugs (AEDs), it has long been speculated that inherited dysfunction of such channels may account for some of the heritability of epilepsy. Indeed, the first mutations discovered to cause epilepsy inherited in a Mendelian fashion were found in genes encoding subunits of voltage- and ligand-gated ion channels.5,7,14,91,99,108,109 Even though monogenic epilepsies are individually rare, they have now been associated with a wide range of ion channel gene mutations. One or two new channel genes responsible for monogenic epilepsy have been reported in each of the last few years (Chapter 18).
Voltage-gated ion channels underlie both the resting and action potentials of neurons. With the exception of the chloride (Cl–) channel CLCN2, which was recently implicated in epilepsy,44 the voltage-gated ion channels that underlie neuronal excitability all contain pore-forming subunits that share a common motif. Indeed, the genes encoding the essential core of ion channels are all thought to be related to a common ancestral prokaryotic potassium (K+) channel, and together comprise the third largest family in the human genome.112 The central feature of this family is a pair of transmembrane helices joined by an extracellular loop that plunges back into the membrane and contributes to the ion-conducting pore. Most K+ channels are composed of four such subunits, so that permeating ions travel through a tunnel lined by four symmetrical peptide chains. Among K+ channels the largest group consists of subunits that have four additional transmembrane helices, making a total of six membrane-spanning segments, with cytoplasmic tails at either end of the peptide chain (Fig. 1). The crystal structure of a representative voltage-gated K+ channel has now been solved67 and is invaluable to efforts to understand the functional impact of missense mutations.
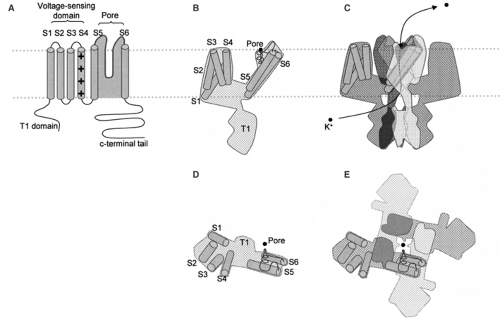 FIGURE 1. Structure of voltage-gated K+ channels. A: A simplified depiction of the peptide chain of a voltage-gated K+ channel showing the six transmembrane segments (cylinders) in the membrane (dotted gray lines). The position of the voltage sensor is indicated (+ signs). The loop between S5 and S6 forms the pore and selectivity filter (shaded gray). The outside of the cell is up, and the intracellular side is down. B: A representation of a single subunit of a voltage-gated K+ channel based on the recent crystal structure.67 The approximate positions of the helices are indicated along with the pore loop and a single potassium ion in the selectivity filter (black circle). The T1 domain is comprised of the N-terminal tail of the subunit and is thought to be important for subunit assembly.85 C: Four subunits arranged to make a channel in the membrane. The helices are only shown in one subunit for clarity. The usual path of a K+ ion through the lower side of the channel, and out the center of the top is indicated. D: A single subunit rotated and viewed from outside the cell, with the positions of the transmembrane domains indicated as in B. E: Four subunits forming the pore of a voltage-gated K+ channel viewed looking down at the surface of a cell. The structure determined by Long et al.67 largely confirmed the membrane topology represented in A, but surprisingly, showed that the voltage-sensing domain (S1–S4) is offset to the side of the pore-forming segments (S5–S6) when the channel is viewed from above the surface of the membrane (looking into the pore of the channel). The black dot represents a K+ ion in the pore of the channel. |
The typical pore-forming subunit of a Na+ or calcium (Ca2+) channel is thought to have evolved via twofold duplication of a six-transmembrane domain (6-TM) K+ channel gene, giving rise to a single protein containing four domains, each of which contains six transmembrane helices (Fig. 2). Although these channels exhibit distinct ion selectivities, they have the same overall topology, pore structure, and fourfold symmetry as the 6TM family of K+ channels. The different ion selectivities exhibited by K+, Ca2+, and Na+ channels are accounted for by a few amino acids in the pore loop. These channels also share a common mechanism of voltage sensing, which involves positively charged amino acids in the fourth transmembrane segment of each subunit (in the case of 6TM K+ channels) or domain (in the case of Na+ and Ca2+ channels). A complete native ion channel also contains one or more auxiliary subunits, which belong to a far more heterogeneous family of proteins, some of which are entirely intracellular while others contain membrane-spanning segments. These modulate the function of the pore-forming subunits, as well as helping to target them
to their correct destination in the neuron and linking them to scaffolding and signaling proteins.
to their correct destination in the neuron and linking them to scaffolding and signaling proteins.
In addition to their different ion selectivities, which relate the direction of ion flux to the chemical and electrical gradients across the membrane, channels have several other important features that account for their distinct roles in neuronal excitability. Notably, they activate (that is, undergo a conformational change that opens the ion-conducting pathway) at different rates and over different transmembrane voltage ranges. Many channels also inactivate in the presence of sustained depolarization, via a conformational change that is not simply the inverse of the activation event. The fast activation and inactivation kinetics of Na+ channels, along with the positive reversal potential of Na+ ions, underlies the upstroke of fast action potentials in almost all mammalian neurons. K+ channels, which tend to hyperpolarize cells when they open, are largely responsible for setting the resting potential of the cells, as well as curtailing action potentials and shaping the frequency and pattern of repetitive firing. Ca2+ channels mediate slow action potentials in some neurons (particularly in the thalamus) but also couple depolarization with intracellular signaling, in the form of Ca2+ entry. One of the most important functions of Ca2+ channels is to trigger neurotransmitter release at synapses.
This overview of voltage-gated ion channels provides the backdrop summarized for further understanding the specialization and diversity of K+, Na+, and Ca2+ channels. Each class may be further subdivided into distinct subtypes, encoded by different genes, different splice variants, and different combinations of auxiliary subunits. This variability produces a functionally diverse population of channels with distinct patterns of expression in brain areas, among neuronal populations, and within cell compartments such as dendrites, axons, and pre- synaptic membranes.
Relating Gain or Loss of Function to Epilepsy
A simplistic view of epileptic channelopathies is that gain-of-function mutations of Na+ or Ca2+ channels (that is, mutations that enhance the Na+ or Ca2+ current flow when the membrane is depolarized) should enhance the excitability of neurons and circuits and therefore lower seizure threshold. Conversely,
loss-of-function mutations of K+ channels should destabilize resting membrane potentials and predispose the brain to seizure. This view is supported, for example, by the fact that several AEDs (phenytoin, carbamazepine, and lamotrigine) are thought to act by stabilizing Na+ channels in an inactivated state, thus rendering them more reluctant to open upon depolarization.81 As will be seen, this view is far from providing a full account of how ion channel mutations give rise to epilepsy.
loss-of-function mutations of K+ channels should destabilize resting membrane potentials and predispose the brain to seizure. This view is supported, for example, by the fact that several AEDs (phenytoin, carbamazepine, and lamotrigine) are thought to act by stabilizing Na+ channels in an inactivated state, thus rendering them more reluctant to open upon depolarization.81 As will be seen, this view is far from providing a full account of how ion channel mutations give rise to epilepsy.
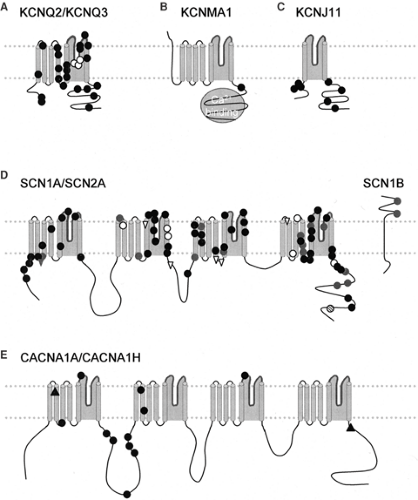 FIGURE 2. Missense mutations associated with epilepsy in voltage-gated channels. A: The subunits underlying the M current, the Kv7.2 and Kv7.3 subunits, encoded by KCNQ2 and KCNQ3, respectively. These subunits share the overall six-transmembrane topology of the archetypical K+ channel shown in FIGURE 1A. Mutations in KCNQ2 are black circles, and KCNQ3 are white circles. (Adapted from Turnbull J, Lohi H, Kearney JA, et al. Sacred disease secrets revealed: the genetics of human epilepsy. Hum Mol Genet. 2005;14(Spec No. 2):2491–2500, with permission.104) B: The BK channel KCNMA1. This channel contains an extra TM domain before the conserved voltage-sensing domains, traditionally called S0. In addition, the channel contains a conserved Ca2+ binding domain (gray circle). The approximate position of the mutation associated with epilepsy is indicated by a black circle. Adapted from Du W, Bautista JF, Yang H, et al. Calcium-sensitive potassium channelopathy in human epilepsy and paroxysmal movement disorder. Nat Genet. 2005;37(7):733–738, with permission.24 C: The approximate positions of missense mutations in KCNJ11 (Kir6.2) linked to DEND syndrome (black circles). The two transmembrane segments KCNJ11 are homologous to the S5 and S6 transmembrane segments in the K+ channel shown in FIGURE 1A. Although KCNJ11 does not have a voltage-sensing domain, it is a member of the voltage-gated channel superfamily and likely shares an evolutionary ancestor with 6TM channels. Functional Kir6.2 channels are associated with the SUR receptor, a multitransmembrane protein. Adapted from Hattersley AT, Ashcroft FM. Activating mutations in Kir6.2 and neonatal diabetes: new clinical syndromes, new scientific insights, and new therapy. Diabetes. 2005;54(9):2503–2513. with permission.43 D: Missense mutations in Na channels linked to epilepsy. The pore-forming subunit of Na channels contains four domains each with six transmembrane segments homologous to those in the 6TM voltage-gated K+ subunit. Missense mutations in SCN1A are associated with SMEI (black circles), GEFS+ (gray circles), intractable childhood epilepsy with generalized tonic–clonic seizures (ICEGTC) (white circles), and infantile spasms (hatched circle). Missense mutations in SCN2A are linked to GEFS+ (gray triangles), and benign familial neonatal-infantile seizures (BFNIS) (white triangles). The two mutations in SCN1B linked to GEFS+ are also shown (gray circles) along with the membrane topology of that subunit. Many further mutations linked to SMEI are nonsense mutations; these are not shown here because they would be likely to trigger NMD, and thus their approximate position in the protein may not be relevant. Adapted from Meisler MH, Kearney JA. Sodium channel mutations in epilepsy and other neurological disorders. J Clin Invest. 2005;115(8):2010–2017, with permission.72 E: Mutations in Ca2+ channels linked to epilepsy. The overall structure of the voltage-gated Ca2+ channels is similar to that of Na+ channels, but conserved amino acid substitutions in the pore lead to selectivity for Ca2+ ions. Missense mutations/polymorphisms in CACNA1H (T-type Ca channel) linked to epilepsy (black circles), from Chen Y, Lu J, Pan H, et al. Association between genetic variation of CACNA1H and childhood absence epilepsy. Ann Neurol. 2003;54(2):239–243, with permission.16 Mutations in CACNA1A (P/Q type Ca channel) linked to epilepsy (black triangles).49,52 The mutation in the intracellular carboxyl tail of the channel is a nonsense mutation, but may escape degradation from the NMD pathway, and is therefore included with missense mutations. |
Currently, missense mutations are the most common type of mutation found in voltage-gated ion channel genes associated with epilepsy. This may however be an artefact of sequencing efforts that focus on coding exons, while ignoring the introns and regulatory sequences within a gene. It may also reflect the potentially more severe impacts of nonsense mutations or mutations that disrupt the regulation of ion channels. Some mutations may not be compatible with fetal viability or with survival to adulthood.
Ion Channel Mutations in Epilepsy
K+ Channels
For mutations of one type of K+ channel, the simple view that relates a reduction in seizure threshold to loss of function appears to hold. Kv7.2 and Kv7.3 subunits, encoded by KCNQ2 and KCNQ3, co-assemble to form a slowly activating K+ channel that shows little inactivation with prolonged depolarization. The subunits are widely expressed in the gray matter of the brain, including γ-aminobutyric acid (GABA)ergic and dopaminergic neurons of the thalamus and basal ganglia.19 The heteromeric channels consisting of Kv7.2 and Kv7.3 have the biophysical properties of the so-called M current,110 which is active in many neurons close to the resting potential and is profoundly modulated by muscarinic and other G-protein–coupled receptors. The molecular identity of this conductance was only resolved with the discovery of the KCNQ2 and KCNQ3 genes. These were identified through a positional cloning effort to elucidate two loci for benign familial neonatal convulsions (BFNC) (see also Chapter 223).7,14,91 Infants affected by this condition have brief generalized and partial seizures, with otherwise normal brain development and behavior; this topic has been reviewed.62 The convulsions generally resolve spontaneously by the age of 6 weeks, although they can persist into adulthood in a minority of cases. All the missense mutations in KCNQ2 and KCNQ3 associated with BFNC impair K+ flux when measured in heterologous expression experiments, mainly by reducing current density.50,92 Loss of function can be expected to apply for the splice site, frameshift, and gene deletion mutations that have also been reported.
These findings underline the importance of the M current for setting the resting potential and firing rates of neurons that have a strategic role in seizure initiation and/or propagation.20,63 Furthermore, the reasonably straightforward link between mutations that decrease the M current and the development of epilepsy has led to renewed interest in the experimental AED retigabine, which relatively selectively increases M currents; two reviews on this topic have been published.20,103 Why infants carrying KCNQ2 or KCNQ3 mutations “outgrow” neonatal convulsions is unexplained. Because the mutations are dominantly inherited, only one allele of one of the two constituent subunits of the heteromeric channels is potentially defective in affected individuals, so there is some degree of redundancy. Moreover, a developmental increase in seizure threshold may occur during the first few weeks of life, or there may be a gradual increase in expression of the remaining, unaffected KCNQ2/3 alleles, such that a small reduction of K+ flux may become insufficient to manifest as spontaneous convulsions. Interestingly, one missense mutation, associated with a broader syndrome that includes myokymia in adulthood, as well as neonatal convulsions, alters a voltage-sensing residue of KCNQ2.23 This mutation produces a slowing of activation that persists upon coexpression of wild-type KCNQ2 and KCNQ3, suggesting a dominant effect on heteromeric channels.
In striking contrast to mutations in KCNQ2 and KCNQ3, mutations found in two other classes of K+ channels associated with epilepsy lead to a gain of function. The KCNMA1 gene, which encodes the pore-forming subunit of a voltage- and Ca2+-sensitive K+ channel, has recently been linked to a syndrome characterized by generalized epilepsy and paroxysmal dyskinesia.24 The large-conductance Big K+(BK) channel encoded by KCNMA1 is widely expressed in the CNS. It contributes to fast neuronal repolarization following action potentials and to the early part of the afterhyperpolarization. It is also present presynaptically, where it may curtail neurotransmitter release. Surprisingly, the KCNMA1 mutation identified by Du et al.24 in a single large family increases the open probability of these channels when expressed in vitro, such that they mediate a larger K+ flux than wild-type channels over a range of transmembrane voltages or intracellular Ca2+ concentrations. A possible explanation for the occurrence of seizures and dys- kinetic paroxysms is that accelerated repolarization allows cells to fire repetitive action potentials. This is consistent with the effect of experimental deletion of an auxiliary subunit in the mouse, which paradoxically enhances BK channel function and is associated with temporal lobe seizures.9
Gain-of-function mutations in the KCNJ11 gene, which encodes the adenosine triphosphate (ATP)-sensitive inwardly rectifying K+ channel Kir6.2, are associated with neonatal diabetes. Although this channel is abundant in the pancreas, where it gates the release of insulin, it is also widely expressed in the brain. Interestingly, some mutations that dramatically reduce the sensitivity of the channel to intracellular ATP (which normally closes the channels) are, in addition, associated with developmental delay, muscle weakness, and epilepsy (the DEND syndrome).37,78,79 Mutant Kir6.2 channels tend to be open at the resting potential, so they are unlikely to lead to neuronal hyperexcitability. Why, then, are they associated with seizures? It has been suggested that ATP-sensitive K+-channel subunits (and the sulfonylurea receptors associated with them in the native channel complex) are expressed to a greater extent in inhibitory interneurons than in pyramidal cells, and the disproportionate inhibition of interneurons by mutant Kir6.2 subunits may lead to seizures; see Ashcroft4 and citations within.
Na+ Channels
Many heterozygous Na+ channel mutations are associated with epilepsy. The first missense mutation identified in a GEFS+ kindred affects the β1 auxiliary subunit encoded by SCN1B (see also Chapter 256). This subunit has a single transmembrane segment, and the mutation disrupts a cysteine bond within the extracellular portion of the protein, preventing it from modulating the function of the pore-forming subunit.109 Although this represents loss of function for the SCN1B gene, the effect on Na+ currents appears to be a net gain of function, because one of the normal roles of β1 is to accelerate fast inactivation. Impairment of fast inactivation represents a striking parallel with mutations in SCN4A, which encodes the pore-forming subunit of the skeletal muscle Na+ channel, associated with hyperkalemic periodic paralysis and paramyotonia.55 Impaired fast inactivation of muscle Na+ channels gives rise to a persistent Na+ current, which leads to depolarization and repetitive firing of muscle fibers.
Several mutations in SCN1A, encoding the pore-forming subunit of the brain NaV1.1 channel, have since been identified in other families with GEFS+27; reviewed in Lerche et al.63 Some of these have also been reported to impair fast inactivation.69 This parallel between muscle and neuronal Na+ channel mutations has, however, been challenged by several observations. First, detailed biophysical examination of several SCN1A mutations reveals effects on activation threshold and other parameters that imply that at least some of them result in loss rather than gain of function.1,68,96 That is, depending on the pattern of membrane potential changes imposed on mutant NaV1.1 channels, they may be more reluctant to open than wild-type channels. Second, disruptive mutations of SCN1A are a frequent cause of severe myoclonic epilepsy of infancy (SMEI, or Dravet syndrome).18,100 This intractable pediatric epilepsy syndrome includes febrile and afebrile generalized and partial seizures, and is associated with episodes of status epilepticus, developmental arrest and regression, and mortality in childhood. It is generally sporadic, although it also occurs in families with a history of FSs. Indeed, an overlap occurs between GEFS+ and SMEI.87 SCN1A mutations associated with SMEI are frequently de novo, and are typically nonsense, splice site, and frame shift mutations, although missense mutations also occur. Many of these mutations are expected to lead to complete loss of function (see later discussion). Finally, missense mutations in SCN1A have also been shown to occur in a milder pediatric syndrome, intractable childhood epilepsy with generalized tonic–clonic seizures (ICEGTC),34 with a variety of effects on activation and inactivation kinetics.83 Notwithstanding the continued uncertainty about whether impaired Na+ channel inactivation found in some GEFS+ mutations outweighs the other kinetic alterations, there is general agreement that the severe end of the GEFS–SMEI spectrum is consistently associated with loss of function SCN1A mutations. In keeping with this, lamotrigine, which promotes Na+ channel inactivation, can aggravate SMEI.40
The controversy surrounding gain versus loss of Na+ channel function in GEFS+ calls for an improved understanding of the normal role of the β1 subunit and of NaV1.1 channels in neuronal excitability. β1 is ubiquitously expressed and co-assembles not only with the pore-forming subunit encoded by SCN1A but also with several other Na+ channels, including NaV1.2 encoded by SCN2A. Interestingly, NaV1.1 and NaV1.2 have complementary expression profiles in many neocortical neurons, with NaV1.1 predominantly expressed in the soma and dendrites, whereas NaV1.2 is mainly located in the axons. Recently, heterozygous SCN2A mutations were identified in association with another pediatric seizure disorder, benign febrile neonatal/infantile convulsions (BFNIC).45
A possible explanation for the association of loss-of-function mutations of Na+ channels with epilepsy is that they are more important for the firing of inhibitory interneurons, although this remains to be tested. Alternatively, Na+ channel mutations might result in unexpected compensatory alterations in the expression of other channels, resulting in a hyperexcitable state. Genetic mouse models may provide an important insight into the consequences of deletion or missense mutations of Na+ channels. The homozygous SCN1B knockout has seizures and ataxia, and exhibits subtle morphologic abnormalities in myelinated axons.15 Interestingly, NaV1.3 expression is increased in some neurons, providing some support for the hypothesis that epilepsy arises from compensatory alterations in other channels. The phenotypes of SCN1A knockin and knockout mice have not yet been reported.
Ultimately, we need to know how Na+ channels behave in situ in people affected by these mutations. Although brain tissue is not accessible to detailed biophysical analysis, Bostock and colleagues have developed transcutaneous stimulation and recording methods that yield an insight into the conductances underlying action potential generation and refractoriness in peripheral nerves.8 Neither SCN1A nor SCN2A are thought to be expressed in peripheral nerve. However, β1 subunits are found in motor axons. A comparison of controls and subjects with the original SCN1B mutation associated with GEFS+ reveals changes that imply a loss of excitability59 consistent with a decreased number of Na+ channels expressed in the nodes of Ranvier, rather than with a change in channel gating.15 This again points to a loss-of-function effect, leaving unexplained the occurrence of seizures if the same phenomenon occurs in the brain.
Ca2+ Channels
T-type Ca2+ channels are unusual among Ca2+ channels in that they activate with relatively small membrane depolarization from resting potentials, but are also readily inactivated with maintained depolarizations. These properties mean that they mediate a transient Ca2+ conductance, revealed by briefly depolarizing neurons from a relatively negative potential. This can be demonstrated in thalamic neurons, which express T-type channels. These cells can be induced to fire rhythmic bursts of Na+ action potentials riding on a slower T-type Ca2+ action potential. As each Ca2+ action potential terminates upon inactivation of the T-type channels, the neuron hyperpolarizes (especially if it receives a concurrent inhibitory synaptic input), following which the cycle repeats itself as the T-type channels recover from inactivation and begin a new Ca2+ action potential. Because of their association with rhythmic burst-firing, T-type channels have long been suspected to play a central role in spike-and-wave epilepsy, which manifests as excessive synchronous oscillations in the thalamocortical loop.
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


