Zonisamide
Masakazu Seino
Ilo E. Leppik
Introduction
After a long hiatus in the development of novel drugs for treating epilepsy, the early 1990s saw several new antiepileptic drugs (AEDs) developed and approved for use in patients with epilepsy. This progress in the treatment of epilepsy may have occurred because we understand more about the mechanisms of action of existing AEDs as well as the underlying pathophysiology of epileptic seizures. However, despite the advances made, a large number of patients with various types of seizures still remain refractory to treatment using existing AEDs.
One of the new drugs to join the global therapy list of AEDs during the 1990s is zonisamide (ZNS) (Fig. 1), a broad-spectrum AED that was developed in Japan by Dainippon Pharmaceutical Co. Ltd. Zonisamide was originally synthesized in 1972 by Uno et al.58,59 as one of a series of 1,2-benzisoxazole derivatives. In the course of routine tests with these derivatives, some of the sulfonamide analogs, most notably ZNS, showed anticonvulsant activity in maximal electroshock (MES) experiments. In animal models, ZNS also demonstrated strong anticonvulsant activity and, based on its structure–activity relationship, was selected as a candidate for development.55
Structure and Chemistry
Zonisamide (1,2-benzisoxazole-3-methanesulfonamide) (Fig. 1) is chemically similar to indole. Zonisamide is synthesized from 1,2-benzisoxazole-3-acetic acid, which is prepared by the Posner reaction between 4-hydroxycoumarin and hydroxylamine.58,59 The intermediate compound, 1,2-benzisoxazole-3-methanesulfonic acid sodium, was synthesized by a chlorosulfonic acid–dioxane complex method. The molecular formula of ZNS is C8H8N2O3S, its molecular weight is 212.23, and its melting point is 164°C to 168°C. Zonisamide consists of nonhygroscopic white to pale yellow crystals or crystalline powder, and it has a slightly bitter taste. It is freely soluble in acetone, sparingly soluble in methanol, and slightly soluble in 95% ethanol. Solubility in water depends on pH and increases sharply at a pH of 8. The infrared spectrum shows absorption bands indicating an amino group at 3,320 and 3,160 cm-1 and a sulfonyl group at 1,330 and 1,145 cm-1. The nuclear magnetic resonance spectrum shows signals corresponding to methylene protons, amino protons, and aromatic protons.59 The ultraviolet spectrum in a methanol solution shows absorption maxima at 239, 245, and 284 nm. The N-O bond of ZNS was cleaved by hydrogenation to give 1-(2-hydroxybenzoyl) methanesulfonamide and its imide, the former being identical to one of the metabolites. At present, plasma and urine concentrations of ZNS can be measured using high-performance liquid chromatography (HPLC),20,35 gas chromatography,35 and enzyme immunoassay21 without disturbance of the other AEDs and their metabolites.
Basic Mechanisms of Action
ZNS was tested in many animal models to investigate its antiepileptic properties. When administered orally, ZNS protected mice, rats, rabbits, and dogs from induced maximal seizures.30 The plasma concentrations that suppressed the tonic extensor components of MES were approximately 10 mg/L in all of the animals examined30,31 and caused neurologic side effects at concentrations over 70 mg/L, demonstrating a wider therapeutic range than phenytoin (PHT) or carbamazepine (CBZ).31 In contrast to the latter two drugs, the anti-MES effect of ZNS remained unchanged after repeated oral administration in rats, indicating no development of metabolic or pharmacodynamic tolerance to its anti-MES effect.32 On the other hand, ZNS had no effect on minimal seizures induced by subcutaneous administration of pentylenetetrazol in mice.30 In spontaneously epileptic rats, ZNS inhibited tonic seizures without affecting absence-like seizures.38 These results suggest that ZNS, like PHT and CBZ, exerts anticonvulsant effects by inhibiting the spread of seizures and has a wider therapeutic plasma concentration range than the latter two drugs.
The antiepileptic action of ZNS was found to result from inhibition of seizure propagation in behavioral studies, and this was confirmed in electroencephalographic (EEG) studies on animal models of epilepsy.18,60 Specifically, ZNS restricted the spread of focal seizures evoked by electrical stimulation of the visual cortex in cats and prevented the propagation of seizures from the cortex to subcortical structures evoked by cortical freezing in cats and by electrical stimulation in visual cortex–kindled cats. One paper reported that ZNS prevented spreading from the amygdala to the hippocampus and cortex of seizures induced by intra-amygdaloid application of kainic acid in rats.54 Other EEG studies confirmed that ZNS has another mode of antiepileptic action, that is, suppression of the epileptogenic focus activity. ZNS, like sodium valproate (VPA), decreased the amplitude and frequency of spikes induced by cortical freezing in cats and abolished interictal spikes induced by cortical application of tungstic acid gel in rats, whereas PHT, CBZ, and phenobarbital did not,13,18,19 suggesting that ZNS suppresses epileptogenic focus activity.
In kindled models, ZNS suppressed clinical convulsions and afterdischarges in cortex- and hippocampus-kindled rats23 and amygdala-kindled rats10 and cats.22 Zonisamide also suppressed photically evoked myoclonus in lateral geniculate–kindled cats.61 In addition to its effectiveness in kindled animal models, ZNS has demonstrated its antiepileptic effects in other animal models of epilepsy.1,17
The mechanism of action of ZNS has been clarified in several cell lines. One of the first reports showed that ZNS selectively increased the time constant for recovery from steady-state slow sodium (Na+) inactivation in voltage-clamped Myxicola giant axon.47 In another report by Rock et al.,46 ZNS, like PHT, blocked the sustained repetitive firing of the intracellular action potential without affecting the initial firing of cultured mouse embryo spinal cord neurons, suggesting an
effect of ZNS on Na+ current inactivation kinetics. Furthermore, Suzuki et al.53 reported that ZNS dose-dependently reduced voltage-dependent transient inward currents (T-type calcium [Ca2+] currents) in cultured neurons of the rat embryo cerebral cortex. This reduction by ZNS of T-type Ca2+ currents was confirmed in cultured human neuroblastoma cells.25 These results suggest that ZNS suppresses neuronal hypersynchronization through the blockage of Na+ and Ca2+ channels.
effect of ZNS on Na+ current inactivation kinetics. Furthermore, Suzuki et al.53 reported that ZNS dose-dependently reduced voltage-dependent transient inward currents (T-type calcium [Ca2+] currents) in cultured neurons of the rat embryo cerebral cortex. This reduction by ZNS of T-type Ca2+ currents was confirmed in cultured human neuroblastoma cells.25 These results suggest that ZNS suppresses neuronal hypersynchronization through the blockage of Na+ and Ca2+ channels.
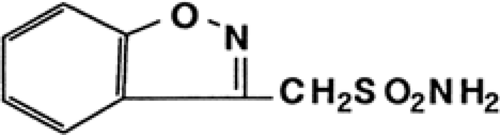 FIGURE 1. Chemical structure of zonisamide. |
Pharmacologic Fundamentals
In animals and humans, ZNS was rapidly and virtually completely absorbed, evenly distributed in the whole tissues including the brain, and rather slowly eliminated. The metabolism of ZNS was extensive, and its excretion route was through urine.
Absorption and Routes of Administration
When [14C]ZNS was administered orally in rats, dogs, and monkeys, radioactivity in plasma reached maximum levels within 3 hours.35 In healthy volunteers administered single oral doses of 200, 400, and 800 mg ZNS, mean plasma concentrations reached peaks of 2.3 to 3, 5.2 to 5.5, and 12.5 mg/L, respectively, within 2 to 5 hours.20,35 The linear pharmacokinetic properties of ZNS were observed in rats and in adult and pediatric patients. When ZNS was administered once a day for 2 days in volunteers, mean peak plasma concentration was 5.1 mg/L after 200 mg daily and 13 mg/L after 400 mg daily. Extrapolation of pharmacokinetic data obtained from single-dose administration indicates that plasma concentration of ZNS reaches steady state after 10 days of daily administration of a 200-mg dose.20
ZNS was markedly concentrated in erythrocytes, as are other sulfonamides. In healthy volunteers administered a 200-mg oral dose, the mean concentration of ZNS in erythrocytes was approximately four to nine times higher and maintained longer than in plasma.20,35 However, uptake of ZNS by erythrocytes was both linear and saturated20—ZNS bound to erythrocytes rapidly at low concentrations, and therapeutic concentrations of ZNS in erythrocytes and whole blood exerted a nonlinear relationship with the dose administered.41 Thus, it is recommended that the plasma or serum concentration of ZNS be measured for therapeutic monitoring. Erythrocytes contain a large amount of carbonic anhydrase. Thus, it is possible that ZNS, like acetazolamide, inhibits the enzyme. However, the inhibition by ZNS of human carbonic anhydrase II, an essential enzyme involved in the antiepileptic effect of acetazolamide, was 180 times less potent than that by acetazolamide.29
Distribution and Protein Binding
Distribution of [14C]ZNS was rather uniform. The levels of radioactivity in most tissues, including brain, were similar to plasma levels, with slightly higher levels in the liver and kidney, and decreased in parallel with the plasma levels in rats.34,35 In pregnant rats, levels of radioactivity in the fetus and placenta were similar to maternal plasma levels.6 Tissue levels in sucklings were virtually the same as in young adult rats.33 When [14C]ZNS was administered intravenously, high uptake was observed in the rat cortex and midbrain.37 In a single-oral-dose study in epileptic patients, the apparent volume of distribution decreased from 1.8 L/kg after a 200-mg dose to 1.2 L/kg after an 800-mg dose, reflecting the nonlinear binding characteristics of ZNS in erythrocytes.20
Protein binding of ZNS was 30% to >50% in rat plasma and serum24,35 and >50% to 60% in human sera.35,41 In human serum, approximately >50% of ZNS bound to protein in vitro at 21.3 mg/L; binding was not significantly affected by the presence of therapeutic levels of PHT, phenobarbital, and sulthiame.35
Metabolism
The metabolism of ZNS was rather extensive, measured by evaluating metabolites in animal urine.36,52,66 Direct acetyl or glucuronyl conjugation occurred. Hydroxylation occurred and was followed by oxidation of the methylene carbon of the sulfamoylmethyl group, finally resulting in loss of the sulfamoyl group. N–O bond cleavage of the isoxazole ring produced two ring-cleft metabolites. Hydroxylation also occurred in the benzene ring. The latter three pathways were followed by conjugation with sulfuric acid or glucuronic acid. Analysis of urine samples in healthy volunteers revealed that ZNS was metabolized by acetylation, and cleavage of the isoxazole ring was followed by conjugation with glucuronic acid.20,35 The P450 CYP3A species is the major isoenzyme involved in its metabolism to the primary metabolite.39 Metabolites of ZNS, with the exception of acetyl-ZNS, were pharmacologically ineffective.
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


