CHAPTER 28 Psychosis and Schizophrenia
OVERVIEW
Schizophrenia was originally described at the close of the nineteenth century by Emil Kraepelin (who categorized madness into episodic mood disorders and chronic psychotic illnesses; he termed the latter dementia praecox, which was later renamed schizophrenia by Eugen Bleuler). In the 100 years since the condition was identified, much progress has been made (in large part the result of the discovery of the first antipsychotic, chlorpromazine), turning schizophrenia from an illness treated in asylums (state hospitals) to one treated in community settings (Figure 28-1).1 However, the fundamental Kraepelinian dichotomy between schizophrenia (and related psychotic disorders) and manic-depressive illness has remained unchanged.
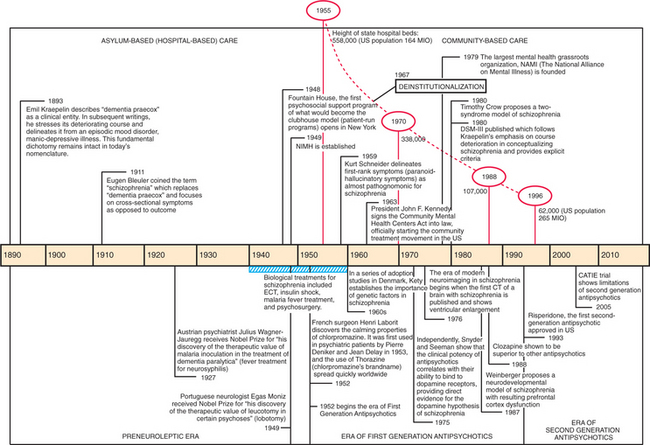
Figure 28-1 Timeline of 100 years of schizophrenia in the United States.
(Source: Shorter E: A history of psychiatry: from the era of the asylum to the age of Prozac, New York, 1997, John Wiley & Sons; Geller JL: The last half-century of psychiatric services as reflected in psychiatric services, Psychiatr Serv 51:41-67, 2000.)
EPIDEMIOLOGY AND RISK FACTORS
Schizophrenia is a syndrome that occurs in all cultures and in all parts of the world. Epidemiological studies have found incidence rates between 7.7 and 43.0 (median 15.2) new cases per 100,000.2 The point prevalence is approximately 5 per 1,000, and the lifetime morbidity risk is approximately 1%. Gender differences exist; males have a 30% to 40% higher lifetime risk for schizophrenia than females,3 and the age of onset is roughly 3 to 4 years later for females.4
The best-established risk factors are not genetic, but environmental (Table 28-1). Immigrant populations have an increased risk,5 with the highest risks for the children of first-generation immigrants, followed by the immigrants themselves.6 Urban living increases risk relative to rural living.7 Etiological insults during brain development include intrauterine infections, particularly influenza,8 and nutritional deficiencies. Maternal starvation has strong support from two epidemiological studies of famines (the Dutch Hunger winter of 1944-19459 and the 1959-1961 Chinese famine10). Both studies found a doubling of risk for schizophrenia in offspring of mothers who starved during the first trimester. Other early environmental risk factors include obstetrical complications.11 Later environmental risk factors are head injury12 and substance use (particularly cannabis).13 Frequent pre-morbid cannabis use (i.e., more than 50 uses) increases the risk for the development of schizophrenia by sixfold.14
Table 28-1 Risk Factors for Schizophrenia
| Nongenetic (Environmental) |
| Prenatal infection (e.g., rubella and influenza) and starvation Obstetric complications (e.g., Rh incompatibility, preeclampsia, and hypoxia) Season of birth (winter birth) Place of birth (urban) Immigration Head injury Drug use (LSD, cannabis, and amphetamines) |
| Genetic |
| Family history (first- and second-degree relatives) Paternal age Genetic syndromes (e.g., VCFS and Klinefelter syndrome) Specific susceptibility genes (see Table 28-2) |
VCFS, Velo-cardio-facial syndrome.
References: 6, 11, 12, 109.
The majority of patients with schizophrenia lack a family history of the disorder. Nevertheless, the fact that genes matter was shown in now-classic twin and adoption studies of the 1960s. For monozygotic twins, the risk of developing schizophrenia approaches 50% for the unaffected twin if the co-twin has schizophrenia. Having siblings or parents (i.e., first-degree relatives) with schizophrenia increases an individual’s risk to approximately tenfold above that seen in the general population. With greater genetic distance, the risk for schizophrenia decreases to a twofold risk over the population risk for second-degree relatives.15
Paternal age increases the risk for schizophrenia in a linear fashion. Compared with the children of fathers who are less than 25 years old, the relative risk for children increases steadily with paternal age, to about 2.0 for fathers in the 45 to 49 age-group, and to almost 3.0 for fathers older than age 50.16 This increase in risk with paternal age is consistent with the hypothesis that de novo mutations contribute to the genetic risk in schizophrenia. New mutations could explain why a disorder with lower fertility rates has not disappeared; in males as opposed to females, the germ-line cells, spermatogonia, continue to divide throughout life, allowing replication errors to accumulate and to be transmitted to offspring.
It is unlikely that a single gene with a significantly large effect will be found that can cause schizophrenia. Most likely, schizophrenia is similar to other non-Mendelian complex disorders where many different genes each make a small, yet important, contribution to disease vulnerability. Such vulnerability genes convey susceptibility without directly causing the disease: the disease is only expressed when combined with other genes or certain environmental factors (e.g., infection or drug use). The number of susceptibility genes is unknown; however, many genes can confer risk in a population. In recent years, several susceptibility genes have been tentatively linked to schizophrenia (for a list of strongest candidates, see Table 28-2).17 The most promising candidate genes are involved in brain development, frontal lobe function, myelination, synaptic function, and glutamate transmission. It is likely that most genes are not specific for schizophrenia; instead, they confer risk for neuropsychiatric disorders that cut across current clinical diagnoses. Several genetic disorders, such as 22q11 deletion syndrome (velo-cardio-facial syndrome [VCFS] or DiGeorge syndrome) or Klinefelter syndrome (XXY syndrome), increase the risk for psychosis in affected individuals.18,19
Table 28-2 Candidate Genes for Schizophrenia
| Gene | Gene Function/Biological Role |
|---|---|
| COMT (catechol O-methyl-transferase) | Functional polymorphism (COMT val allele) increases dopamine catabolism in frontal lobe, resulting in impaired prefrontal physiology110 |
| NRG1 (Neuregulin 1) | Growth factor with pleomorphic role in brain development and function111 |
| Dysbindin or DTNBP1 (dystrobrevin binding protein 1) | Associated with negative symptoms112; involved in synaptic function |
| DISC1 (disrupted in schizophrenia 1) | Interacts with key proteins in signalling pathways and neuronal migration113 |
| RGS4 (regulator of G-protein signalling 4) | Modulator of intracellular signaling for G-protein coupled receptor, including the dopamine receptor114 |
| GRM3 (metabotropic glutamate receptor type 3 gene) | Involved in glutamatergic neurotransmission and prefrontal function115 |
| G72 (G72/G30 gene complex) | Indirectly affects glutamatergic neurotransmission, possibly via interaction with d-amino acid oxidase (DAAO)116 |
Gene selection based on reference 17.
An interaction between susceptibility genes and environment forms the basis for the neurodevelopmental hypothesis of schizophrenia in which a clinically silent, latent propensity toward schizophrenia (e.g., a genetic vulnerability or insult during brain development, such as intrauterine infections or maternal starvation) gets uncovered when the brain matures (e.g., during the naturally occurring pruning of dendritic arborization) or additional insults (e.g., cannabis use) push a vulnerable brain toward psychosis.20 Since the clinical picture (phenotype) is not solely determined by the gene sequence (genotype), but rather by which genes are expressed (the epigenotype), some gene-environment interactions may occur via epigenetic modifications of the genome. Functionally, disconnection between frontal and hippocampal regions has been suggested to account for the emergence of clinical symptoms in this developmental model.
PATHOPHYSIOLOGY
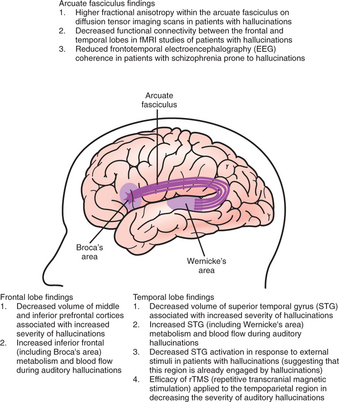
Evidence from neurochemistry, cellular neuropathology, and neuroimaging studies supports the idea that schizophrenia is a brain disease, that is, a disease manifest by abnormalities in brain structure, brain function, or both. The observed abnormalities are thus far of limited specificity and sensitivity, and have therefore not yet had clinical relevance for diagnosis and treatment, and only limited value in prognostication. Furthermore, there is no single universally accepted theory regarding the brain dysfunction seen in schizophrenia, but rather there are a host of competing and overlapping models. (For an example of a model of auditory hallucinations that combines neuroanatomical and neuroimaging findings, see Figure 28-2.)
Neurochemical Changes
The idea that an imbalance in internal chemistry may result in insanity (a notion prevalent since the humoral theories of antiquity) found a modern expression in the “dopamine hypothesis” of schizophrenia.21 The dopamine hypothesis was built on two pillars of evidence: (1) amphetamines, known dopamine receptor agonists, can produce a schizophrenia-like state in healthy adults; and (2) the discovery that the antipsychotic effect of the phenothiazines was associated with their ability to block the D2 dopamine receptor. Further work established the tight relationship between the clinical potency of antipsychotic medications and their affinity for D2 receptors. This confluence of findings suggested that schizophrenia was associated with a hyperdopaminergic state in the mesolimbic system, ameliorated through the use of antipsychotic medication. Modern neuroimaging approaches (such as positron emission tomography [PET] and single-photon emission computed tomography [SPECT]) have directly demonstra-ted heightened dopamine synthesis and presynaptic release in response to use of methamphetamine in patients with schizophrenia.22
While the dopamine hypothesis remains central to our understanding of the therapeutic action of antipsychotics, hyperdopaminergia seems to explain only the psychotic aspect of schizophrenia.23 To account for the lack of therapeutic action of antidopaminergic drugs on other symptom clusters of schizophrenia, particularly negative and cognitive symptoms, a hypodopaminergic state was postulated in which there is a lack of stimulation of prefrontal dopamine-1 neurons.24 In addition, models for schizophrenia involving neurotransmitter systems other than dopamine have been proffered. The impetus for a “glutamate hypothesis” of schizophrenia (with hypofunction of the glutamate system) stems largely from the psychosis-inducing effects of two glutamate antagonists, phencyclidine (PCP) and ketamine.25 Given the ubiquity of N-methyl-d-aspartate (NMDA) receptors and the link between NMDA hyperactivity and excitotoxicity, simply enhancing glutamate release is not feasible, and increasing activity at the glycine modulatory site on the NMDA receptor, or increasing activity at the glutama-tergic a-amino-3-hydroxy-5-methylisoxazole-4-propionic acid (AMPA) receptors, is pursued instead. Acetylcholine, particularly in its actions at intracerebral nicotine receptors, has also received recent attention.26 This relates to the fact that an extraordinarily high percentage of patients with schizophrenia smoke tobacco, consistent with a “self-medication” hypothesis regarding nicotine use. In addition, nicotine appears to have salutatory effects on attention and other cognitive domains know to be affected in these patients. Early work on the development of nicotinic-agonist treatments for schizophrenia is now underway.
Neurocellular Changes
While a unifying neuropathological explanation in the form of a schizophrenic equivalent of the “plaques and tangles” of Alzheimer’s dementia is currently absent, modern stereomorphometric research has uncovered a handful of more subtle abnormalities, primarily within the prefrontal cortex, anterior cingulate gyrus, and medial temporal lobe. There is no clear loss of cortical neurons; instead there is subtle disarray in cortical cytoarchitecture and a decreased volume of neuropil (comprised of axodendritic processes, glia, and cerebral vasculature), suggesting problems during brain development and regional connectivity, respectively.27,28 One relatively specific abnormality of one cell type, a dysfunction of GABA-ergic chandelier interneurons, has been fairly well established.29,30 Any disruption of inhibitory GABA-ergic interneurons in cortical areas would have widespread implications for brain function.31 Another cell type with disrupted function in schizophrenia is the oligodendrocyte; however, the evidence for this is more preliminary.32
Changes in Brain Structure
From the very first images of the human brain, first indirectly by the now-outdated pneumoencephalography, later directly by using computed tomography (CT), one major finding emerged: patients with schizophrenia had larger ventricular volumes than did matched sets of non–mentally ill controls.33 The impact of this finding, which has subsequently been replicated many times using magnetic resonance imaging (MRI), cannot be overstated, because for the first time there was visual evidence of schizophrenia as a brain disease (Figure 28-3). There were (and are) two major limitations to this much-replicated finding: first, ventricular enlargement is neither specific nor sensitive enough to use as a diagnostic tool; second, ventricular enlargement is neuroanatomically vague, making it nearly impossible to link this change with the psychotic phenomena seen clinically.
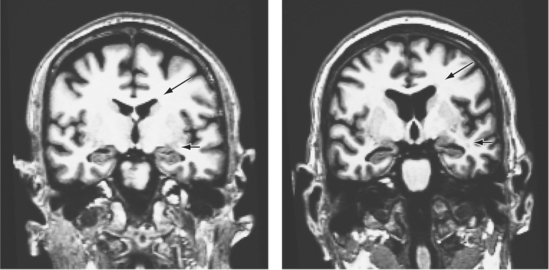
Subsequent work has attempted to find structural differences within the cerebral cortex and the subcortical nuclei. Although structural differences have been observed in a number of areas across the cortical mantle,34,35 the hippocampus stands out as a region where there have been well-replicated findings. Meta-analyses of classical volumetric analyses of imaging data indicate that on average, hippocampal volumes are 4% smaller in patients with schizophrenia than in matched controls.36 Similar volumetric changes within the medial temporal lobe have been confirmed from a meta-analysis of studies using the newer voxel-based morphometric approach.37 Some of the volume changes might occur concurrent with the development of psychosis.38 Using diffusion tensor imaging (DTI), it can be shown that the integrity of white matter tracts is compromised in schizophrenia, consistent with disconnectivity between brain regions.39
Changes in Brain Function
Event-related electroencephalography (event-related potentials [ERPs]) and functional magnetic resonance imaging (fMRI) are technologies used to examine brain function as opposed to brain structure. Using the analysis of ERPs, several abnormal physiological effects, sometimes called endophenotypes, have been well replicated in patients with schizophrenia and may eventually provide the best chance of becoming a clinically suitable diagnostic tool.40 The P50, for example, is an ERP waveform that measures the ability to suppress irrelevant information (sensory gating). P50 is often abnormal, not only in patients with schizophrenia, but also in their clinically healthy relatives. This endophenotype is linked genetically to schizophrenia and to genetic polymorphisms of the alpha-7 nicotinic receptor, a possible treatment target for schizophrenia.41 Another measure of sensory gating, prepulse inhibition (PPI) has been linked to haplo insufficiency of the Tbx1 gene. This gene is one of the affected genes in the 22q11 deletion syndrome that increases the risk for schizophrenia.42
Using fMRI, which produces excellent spatial resolution compared with electroencephalogram (EEG) measures, a number of areas (including the hippocampus and anterior cingulate gyrus) have demonstrated aberrant activation in patients with schizophrenia. However, the preponderance of research has focused on the task-related activity of the prefrontal cortex.43 Although early studies were suggestive of a pattern of task-related hypofrontality, subsequent studies were more consistent with hyperfrontality. This discrepancy appears to be linked to inappropriate modulation of frontal activity in response to varying the nature of the task difficulty.44
CLINICAL FEATURES AND DIAGNOSIS
The diagnosis of schizophrenia is made clinically, based on a typical combination of symptoms (present cross-sectionally and longitudinally), in the absence of other psychiatric or medical conditions that would explain the symptoms (see below). The exact number and combination of symptoms, as well as the required duration of symptoms to make a diagnosis of schizophrenia, differ depending on the classification system used (Table 28-3). Making a diagnosis based on clinical symptomatology and course alone, without the help of genetic markers or biomarkers, can lead to different diagnoses over time (even in the same patient if the clinical picture changes).
Table 28-3 Key Diagnostic Features in Schizophrenia in DSM-IV and ICD-10
| ICD-10 | DSM-IV |
| Active-Phase Symptoms | |
| One characteristic symptom from this list: | One characteristic symptom from this list: |
| Thought echo, insertion, withdrawal, broadcasting* | Typical hallucinations (running commentary, conversing voices) |
| Delusions of control, influence, passivity; delusional perception* | |
| Typical hallucinations (e.g., running commentary, conversing voices)* | |
| Culturally inappropriate and completely impossible delusions | Bizarre delusions |
| OR | OR |
| Two symptoms from this list: | Two symptoms from this list: |
| Other hallucinations with delusions or overvalued ideas | Delusions |
| Hallucinations | |
| Significant formal thought disorder | Disorganized speech |
| Catatonia | Disorganized behavior or catatonia |
| Negative symptoms | Negative symptoms |
| Duration of Symptoms | |
| 1 month of acute symptoms | 6 months of illness (including prodrome); 1 month of acute symptoms |
| Functional Decline | |
| Not required | Required |
| Exclusion Criteria | |
| Psychosis occurs only in presence of significant mood disorder | Schizoaffective disorder, depression with psychotic features |
| Alcohol- or drug-related psychosis | Substance use disorders |
| Organic brain disease | Medical conditions |
| Pervasive developmental disorder (unless psychosis is prominent) | |
| Subtypes | |
| Paranoid (mainly delusions and hallucinations) | Paranoid |
| Hebephrenic (prominent thought disorder) | Disorganized |
| Catatonic | Catatonic |
| Undifferentiated (syndromal but mixture) | Undifferentiated |
| Residual (no syndromal symptom severity) | Residual |
| Simple (insidious onset of only negative symptoms) | — |
| Postschizophrenic depression | — |
Based on references 117 and 118.
* These hallucinatory-paranoid symptoms are based on Schneiderian first-rank symptoms.
The onset of schizophrenia can be acute (i.e., symptoms develop over a few days) or subacute (i.e., symptoms develop over a month), although a more insidious onset with signs of the illness beginning many months or even years before frank psychosis is usually seen (Figure 28-4). A nonspecific prodromal phase can often be ascertained retrospectively. Pro-dromal symptoms include attenuated psychotic symptoms (e.g., suspiciousness, perceptual distortions, or perplexity), depression and suicidality, obsessive thinking, and sleep problems. Role failure and loss of social competence are typical features.45 However, the progression to schizophrenia for patients in a putative prodromal state is not inevitable, with one study reporting a 1-year conversion rate of 41%.46 Unfortunately, even after the development of clear-cut psychosis, the patient and his or her family and friends often do not recognize it. Even when recognized, the affected individual often resists treatment. This results in untreated psychosis that lasts on average almost 2 years.47 The duration of untreated illness, which includes the prodromal period and the duration of untreated psychosis (DUP), can last several years, and typically results in disrupted psychological and social development and impaired role function. The prodromal period is an area of active research, since the functional impairment in cognitive decline seen in schizophrenia occurs before the onset of psychosis and remains stable even after recovery from psychosis.48 Exactly when the cognitive decline begins remains uncertain, but early signs of the disturbance (as judged by scholastic records) point toward onset in middle school or even earlier, possibly with further worsening around the time of psychosis.49
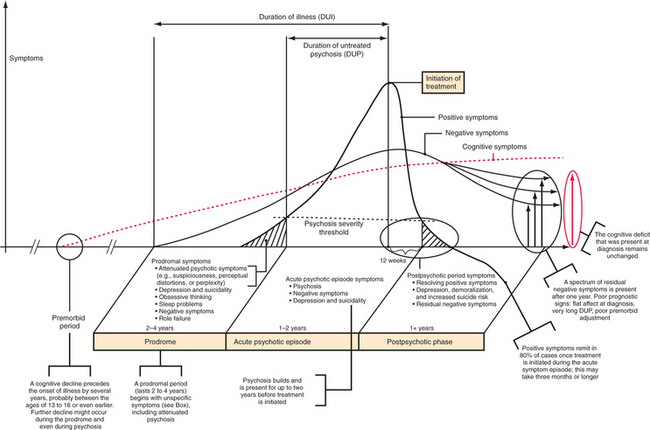
The hallmarks of acute schizophrenia are hallucinations and delusions, sometimes grouped together as positive symptoms. Hallucinations are perceptions without an external stimulus, and they can occur in any sensory modality (Table 28-4). However, by far the most common type of hallucination is auditory, occurring in at least two-thirds of patients over the course of their illness. Certain types of third-person (Schneiderian) hallucinations are frequently encountered: several voices talking about the patient, often in a derogatory way; a voice giving a running commentary on what the patient is doing; or a voice repeating what the patient is thinking. While hallucinations in other modalities are possible, visual or olfactory hallucinations in particular should raise the suspicion for an organic etiology of the hallucinations.
Table 28-4 Types of Hallucinations by Modality in Schizophrenia and Other Conditions
| Visual Hallucinations |
| Visual hallucinations can be simple or elementary (unformed, e.g., a flash of light) or complex (e.g., a face, an animal). While possible in schizophrenia, visual hallucinations (and illusions) are much more typical for and common in deliria and dementias. Other nonpsychiatric etiologies include migraines and seizures, ocular pathology (including the Charles Bonnet syndrome in visually impaired people), narcolepsy, sleep deprivation, and midbrain pathology (peduncular hallucinations). |
| Auditory Hallucinations |
| Auditory hallucinations can be any sound, such as banging doors, footsteps, music, or voices. Common in schizophrenia are so-called Schneiderian hallucinations, characterized by the voice(s) using the third person when talking about the patient (“He is such a loser.”). Depressed patients who hallucinate often experience voices in the self-accusatory second person (“You are such a loser.”). Command hallucinations tell a patient to do a certain act and can include homicide, suicide, or self-mutilation. |
| Olfactory Hallucinations |
| Olfactory hallucinations are a common symptom in temporal lobe epilepsy. If olfactory hallucinations occur in schizophrenia, the hallucinated smells often have the character of a stench (feces, vomitus), but pleasant smells (e.g., perfume) are also experienced. |
| Tactile Hallucinations |
| Tactile hallucinations involve the sensation of being touched or of insects crawling on the skin (formication). This is a typical symptom of cocaine or amphetamine use. Tactile hallucinations occur as a fairly isolated symptom in Ekbom’s syndrome (delusional parasitosis), where the tactile hallucination is elaborated in a delusional way. It also occurs in alcohol and benzodiazepine withdrawal. |
| Somatic Hallucinations |
| Somatic hallucinations involve a sensation arising from within the body; they are fairly common in schizophrenia and are obvious if bizarre (e.g., experiencing movements of the brain). Somatic hallucinations need to be differentiated from symptoms of an as yet undiagnosed disease and from hypochondriacal preoccupation with normal body experiences (e.g., palpitations or bowel peristalsis). |
| Gustatory Hallucinations |
| Gustatory hallucinations are very rare in schizophrenia, but they can occur as part of persecutory delusions (e.g., tasting poison in food). |
Delusions are false, nonculturally sanctioned beliefs that are held with great conviction, even in the face of overwhelming evidence to the contrary. Table 28-5 outlines common delusional themes. The delusional idea puts the patient at odds with his or her culture or subculture. The content of delusions can be nonbizarre (feasible) or bizarre (impossible based on the laws of physics). By convention, bizarre delusions suggest schizophrenia and exclude delusional disorder.
Table 28-5 Types of Delusions by Content in Schizophrenia and Other Conditions
| Persecutory |
| A person or force is interfering with the patient, observing the patient, and wishing to harm the patient (e.g., poisoning him or her). These can be family members, co-workers, government agencies (e.g., the CIA or FBI), or even aliens or the devil. Very common theme in delusional disorder or schizophrenia. |
| Reference |
| Random and innocuous events (e.g., a news item in the morning newspaper, an incidental comment or gesture by a stranger) take on personal significance and meaning. |
| Control |








