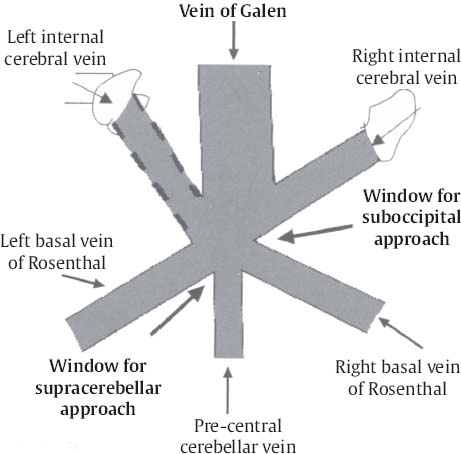
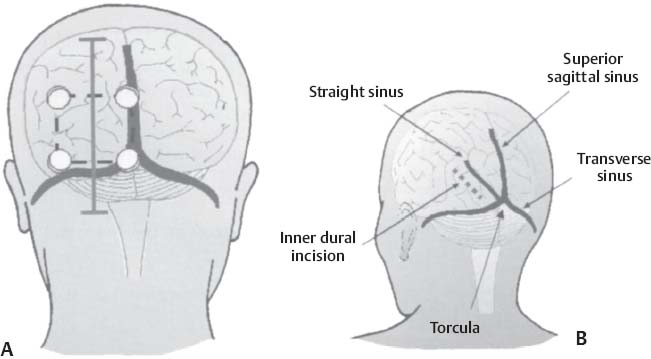
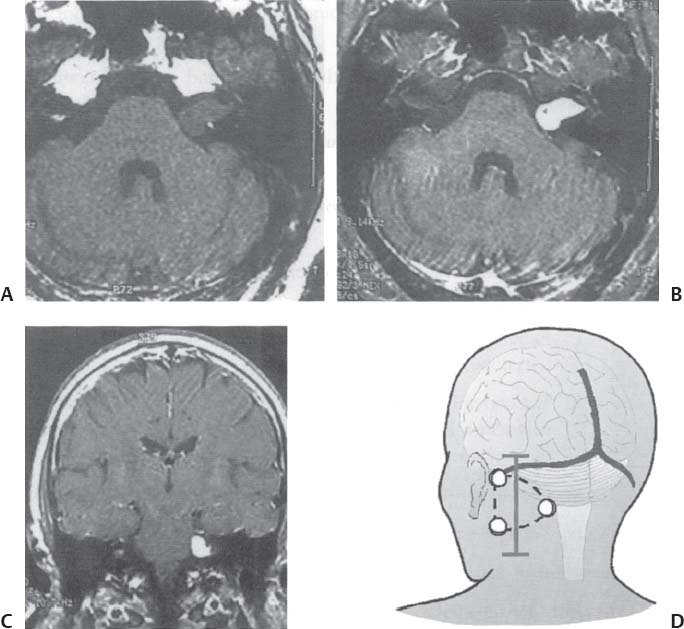
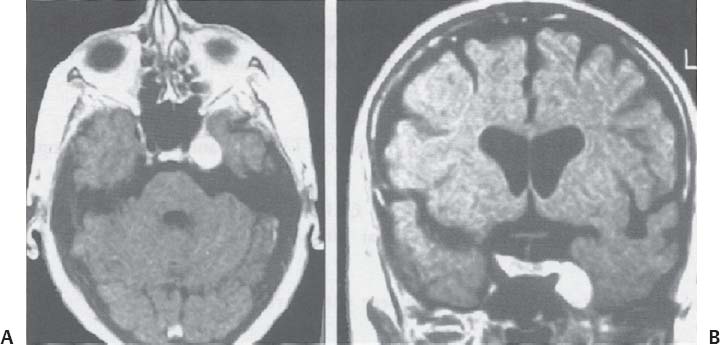
C H A P T E R 8 BRAIN TUMORS I. LOW-GRADE ASTROCYTOMA (WHO GRADE 1 OR 2) A. Treatment 1. Usually with surgery alone (curative if juvenile pilocytic astrocytoma) 2. Radiation dose, if used—45 Gy to the tumor with a 2-cm margin; hold off if young due to long-term side effects 3. Chemotherapy—procarbazine, CCNU, and vincristine (PCV) II. HIGH-GRADE ASTROCYTOMA (WHO GRADE 3 OR 4) A. Prognosis—depends on age (younger is better), Karnofsky score, and histology B. Treatment 1. Cytoreductive surgery—image-guided surgery suggested 2. External beam radiation—typically 60 Gy at 2 Gy fractions 5 days a week, and may add boost to resection cavity 3. Temodar—an oral alkylating agent that increases median survival— from 12 to 14 months when given concurrently with radiation therapy 4. Gliadel (carmustine [BCNU]) wafers—increase survival by 8 weeks 5. Avastin—an anti-VEGF agent may increase survival minimally 6. PCV chemotherapy 7. Surgery—avoid if tumor is bilateral, or in patients with low Karnofsky score C. Survival 1. Per grade—1 (10 years), 2 (8 years), 3 (2 years), and 4 (1.5 year) 2. Average—17 weeks with biopsy, 30 weeks with surgery alone for grade 4 III. OLIGODENDROGLIOMA A. Treatment—surgery, chemotherapy (PCV helps significantly), radiation therapy (XRT; for the anaplastic variety) B. Survival—30% at 10 years; 1p19q loss predicts better chemotherapy response and longer survival IV. EPENDYMOMA A. Evaluation—evaluate for drop metastasis (mets) with lumbar puncture (LP) and spinal magnetic resonance imaging (MRI) B. Treatment 1. Surgery 2. XRT—50 Gy to the tumor bed, and spinal XRT if drop mets are seen or cells detected in the cerebrospinal fluid (CSF) C. Survival—tumor is usually very radiosensitive; survival is 40% at 5 years in children, 80% at 5 years in adults V. CHOROID PLEXUS TUMORS A. Treatment—surgery only; consider preoperative embolization VI. PRIMITIVE NEUROECTODERMAL TUMOR (PNET) A. Evaluation—evaluate for drop mets with spinal MRI and LP B. Treatment 2. XRT—radiate craniospinal axis after resection, after 3 years old in children 3. Surgical resection for medulloblastoma a. Craniospinal XRT—40 Gy with 15 Gy to the tumor bed and drop mets over 6 weeks b. Chemotherapy—CCNU and vincristine 4. Shunt—required in 30% of cases C. Survival—tumor is usually very radiosensitive; survival is 43% in 10 years. VII. GANGLIOGLIOMA A. Treatment—surgery alone VIII. PINEAL TUMORS—pineal region borders are the tectum, vermis, splenium of the corpus callosum (posteriorly), third ventricle (superiorly), and thalami (laterally). A. Evaluation 1. Evaluate for drop mets with all varieties 2. Tumor varieties a. Germ cell—most common; most often germinoma b. Pineocytoma/pineoblastoma, astrocytoma, embryonal carcinoma, choriocarcinoma, and teratoma 3. CSF for α fetal protein (AFP)—in embryonal carcinoma, endodermal sinus tumor, and teratoma 4. BHCG—choriocarcinoma and germinoma B. Treatment 1. If tumor resection does not relieve hydrocephalus—place ventriculoperitoneal (VP) shunt or perform third ventriculostomy (penetrate floor of third ventricle anterior to mamillary bodies). Fig. 8.1 Surgical windows around the deep venous anatomy for pineal region lesions. (With permission from Citow JS. Neurosurgery Oral Board Review. 1st ed. New York, NY: Thieme Medical Publishers; 2003: 60, Fig. 9.1.) 2. Avoid performing a stereotactic biopsy due to the hazard of adjacent vessels unless you are comfortable with third ventricular biopsy 3. Germinoma may be treated with biopsy followed by radiation and chemotherapy. 4. Resection carries a 10% mortality. C. Operative approaches 1. Infratentorial / supracerebellar a. Not as easy with a steep tentorium b. Usually safe to divide the precentral cerebellar vein c. Operative window—between the two basal veins of Rosenthal (Fig. 8.1) 2. Suboccipital/transtentorial a. Risk of visual loss Fig. 8.2 (A) Suboccipital and (B) transtentorial incision. (With permission from Citow JS. Neurosurgery Oral Board Review. 1st ed. New York, NY: Thieme Medical Publishers; 2003: 61, Fig. 9.2A, 9.2B) b. Best for tumors above the tentorial edge and vein of Galen (Fig. 8.2A,B) c. Incise the tentorium 1 cm lateral to the straight sinus d. Operative window—between internal cerebral vein and basal vein of Rosenthal 3. Transcallosal—minimalize to avoid disconnection syndrome 4. Transventricular—through the nondominant superior temporal gyrus IX. PITUITARY ADENOMA A. Apoplexy 1. Signs/Symptoms—typically presents with headache and ophthalmoplegia due to tumor infarction or hemorrhage 2. One third of prolactin-secreting adenomas will hemorrhage. 3. Not an emergency if asymptomatic 4. Treatment in cases with mass effect—steroids and surgical decompression 1. Bitemporal hemianopsia 2. Cavernous sinus invasion—impaired cranial nerves (CNs), chemosis, proptosis, and rarely cerebrovascular accident (CVA) 3. Hypo- or hyperpituitarism a. Increased prolactin (rule out Hook effect) i. Amenorrhea, galactorrhea, and impotence ii. May also be seen with hepatic or renal disease, phenothiazines, verapamil, cimetidine, and pregnancy b. Increased adrenocorticotropic hormone (ACTH)—increased serum and urine cortisol, hyperpigmentation (Nelson syndrome), hypertension, hyperglycemia, abdominal stria, moon face, truncal obesity, extremity wasting, bruising, osteoporosis, amenorrhea, peripheral neuropathy, and depression c. Decreased ACTH—orthostatic hypotension and fatigue d. Increased growth hormone (GH) i. Increased somatomedin C—synthesized in the liver and produces IGF-1 ii. Acromegaly—coarse facial features, enlarging head, hands, and feet iii. Peripheral neuropathy, cardiomyopathy, hypertension, glucose intolerance (diabetes), upper airway obstruction, perspiration, colon cancer, and giantism (if occurs before growth plate closes) iv. Somatostatin decreases GH release. e. Decreased GH—dwarfism f. Decreased thyroid stimulating hormone (TSH)—cold intolerance, fatigue, coarse hair, peripheral neuropathy, and myxedema coma g. Decreased follicle-stimulating hormone/luteinizing hormone (FSH/LH)—amenorrhea, decreased libido, and infertility (hypogonadism) h. Decreased antidiuretic hormone (ADH)—diabetes insipidus (very rare with a sellar mass) C. Evaluation 1. Endocrine history (as above), visual fields, extraocular movements 2. Prolactin—mild elevation to 25–150 is usually by stalk effect from decreased dopamine that normally inhibits prolactin 3. Morning cortisol, 24-hour urine cortisol 4. ACTH—decreased with adrenal tumor, increased with pituitary adenoma or ectopic lung mass 5. Dexamethasone suppression test a. Low dose of 1 mg—normally decreases ACTH b. High dose of 8 mg—decreases ACTH with Cushing disease (pituitary adenoma) c. There is no suppression with an ectopic tumor 6. TSH, T4, GH, somatomedin C, FSH/LH, estrogen, testosterone 7. MRI a. May be normal in 50% of cases b. Enlarged pituitary gland on MRI i. May be caused by pregnancy (also with amenorrhea and increased prolactin) ii. May be caused by primary hypothyroidism (elevated TSH, treat with Synthroid) 8. Petrosal sinus sampling—has been used to evaluate the location of an ACTH-secreting tumor; however, normal people have different amounts of ACTH on each side. 9. Rapid-sequence infused MRI—may demonstrate location of a small adenoma D. Treatment 1. Prolactinoma a. Bromocriptine (Parlodel)—dopamine agonist that decreases tumor size 75% by 8 weeks b. Lifelong therapy needed to control tumor growth c. Starting dose—2.5 mg orally three times daily (PO t.i.d.) d. Initial bothersome side effects—emesis and postural hypotension; normally resolve over several weeks e. Watch for low estrogen—may cause osteoporosis f. Safe to take oral contraceptives (OCP) with bromocriptine and safe to take bromocriptine while pregnant g. If tumor enlarges during pregnancy, surgically resect it or increase the bromocriptine h. Follow patient with visual field examinations i. Dostinex is another medical option. j. Consider transsphenoidal or subfrontal resection if medical therapy fails. 2. Acromegaly a. Surgical resection—best option, followed by therapy with bromo criptine or octreotide, and finally XRT if GH levels do not normalize b. Postop steroid withdrawal is a good sign. c. IGF-1 levels usually drop 4 weeks after surgery. d. Tumors with growth hormone levels > 40 are difficult to cure. 3. Cushing disease a. Surgical resection—best option b. Ketoconazole may be used to temporarily lower cortisol levels. c. Bilateral adrenalectomy—last resort (patient may develop Nelson syndrome) 4. Hyperthyroidism—surgical resection favored over octreotide 5. Radiation b. 50% decrease in ACTH, FSH/LH, and TSH in 10 years c. Visual loss may occur. d. Consider only if unable to reoperate 6. Surgery—subfrontal or transsphenoidal a. Transnasal transsphenoidal—most common approach i. Insert a throat pack, prep the thigh or abdomen for a fat graft, inject the nasal mucosa with epinephrine, enter the right nostril ii. In a submucosal plane, dissect the left side of the nasal cartilage and bilateral nasal bone to the sphenoid bone (find the sphenoid ostium posterior to middle/superior turbinates) iii. Insert the Cushing speculum and finally the Hardy speculum, assess the midline (sphenoid ridge may be off-center), chisel off the anterior wall of the sphenoid sinus, remove the mucosa iv. Chisel off the anterior wall of the sella, incise the dura in cruciate fashion, and curette out the soft tumor v. A newer approach is to insert the speculum directly to the floor of the sphenoid sinus, open the speculum to displace the nasal bone, and open the mucosa over the sinus wall and proceed from there. vi. A lumbar drain may be inserted and air injected to help push the tumor down during resection. vii. Postop MRI should be done at 3 months. E. Complications 1. Diabetes insipidus (DI), injuries to CNs II–VI, carotid artery injury, hypopituitarism, and CSF leak. 2. Antibiotics—use until the packs are removed at 3 days (chloramphenicol and ampicillin) as well as a hydrocortisone stress dose followed by a replacement dose (100 mg intravenously every 8 hours [IV q8h] × 3 then 20 mg PO every morning and 10 mg every bedtime [qHs]). 3. Hydrocortisone—stop 2 days after surgery and send a urine cortisol (< 9 needs steroid replacement). 4. Diabetes insipidus—diagnosed as a urine output (UOP) > 250 mL/h × 3 hours with specific gravity (SG) < 1.005. 5. Lost fluid—replace with ½ normal saline and use 0.1 μg desmopressin acetate (DDAVP) IV q6h or intranasal 10–40 μg twice a day (b.i.d) for long-term maintenance 6. CSF leak—treat initially with a lumbar drain and if this is unsuccessful, consider exploration and packing. 7. Carotid injury with pulsatile bleeding—should be treated with packing followed by an angiogram to determine if bypass or endovascular treatment (occlusion, stenting, etc.) is needed. X. CRANIOPHARYNGIOMA A. Treatment 1. Perform preop and postop endocrine evaluation 2. Steroids—use as in Section X.E.3 after surgery. 3. Subtotal resection—appears to be the best treatment (unless complete removal can be safely achieved), followed by XRT 4. Surgical approach—usually subfrontal 5. Surgical mortality—10%; morbidity includes hyperthermia, lethargy, impaired thirst mechanism, and endocrine deficiencies B. Survival—5-year rate is 75%. XI. COLLOID CYST A. Treatment 1. If < 7 mm and without hydrocephalus, consider conservative management with follow-up MRI at 6 months 3. Surgical pitfalls—damage to anterior cerebral artery, cingulate gyrus, fornix, internal cerebral veins; enter wrong ventricle; intraventrical hemorrhage XII. MENINGIOMA A. Treatment 1. Surgical resection—consider if symptomatic a. Preop—remember to consider dural patch material (autograft or synthetic), dural sinus patency, and possibility of embolization (may cause distant strokes). b. Recurrence with partial resection—60% at 10 years without XRT; 30% with XRT. B. Survival—91% at 5 years XIII. VESTIBULAR SCHWANNOMA A. Evaluation—should include pure tone audiogram and speech discrimination test (50/50 is considered serviceable hearing with 50 Db pure tone hearing and 50% speech discrimination). B. Treatment—if asymptomatic, consider following with MRI evaluation every 6 months (Fig. 8.3A,B) 1. Suboccipital retrosigmoid resection 2. Middle fossa approach—for small intracanalicular tumors distal in the canal 3. Translabyrinthine approach—sacrifices hearing 4. XRT/stereotactic radiosurgery—14 Gy dose, undetermined indication with primary or recurrent tumors, and associated with higher hearing preservation and decreased risk of facial palsy 5. Use intraop 8th (brainstem auditory evoked response [BAER]), 7th, and 5th nerve monitoring. Fig. 8.3 (A) Vestibular schwannoma, noninfused, (B) infused, and (C) axial and infused coronal T1-weighted magnetic resonance images demonstrating an enhancing mass emanating from the left internal acoustic meatus. (D) Vestibular schwannoma exposure via suboccipital–retrosigmoid craniotomy. (With permission from Citow JS. Neuropathology and Neuroradiology: A Review. New York, NY: Thieme Medical Publishers; 2001: 105, Fig. 147.) 6. Suboccipital retrosigmoid craniotomy a. Place the patient semiprone (45 degrees) or lateral with the falx parallel to the floor. b. Incision is the length of the pinna and located 2 cm posterior to the mastoid notch. c. A burr hole is made at the asterion and a small craniotomy fashioned after eggshelling edge of transverse and sigmoid sinuses. d. Dura is opened with triangular flaps based on the transverse and sigmoid sinuses. e. Cerebellum is gently and slowly retracted medially to expose the CNs. f. Water-tight dural closure is essential. g. Complications i. Facial palsy—if unable to close eyes, use natural tears q2h, Lacri-Lube, and tape eyes shut qHs ii. Tarsorrhaphy—if after 1 year the weakness hasn’t improved, perform a XII–VII anastomosis (or even after only 2 months if the nerves were directly cut) iii. CSF leak—treat with a lumbar drain, shunt, or ENT (ear, nose, and throat) packing iv. Vertigo—resolves with rehab C. Preservation of function 1. 7th nerve—95% (< 1 cm), 80% (1–2 cm), 50% (> 2 cm) 2. 8th nerve—57% (< 1 cm), 33% (1–2 cm), 6% (> 2 cm) XIV. TRIGEMINAL SCHWANNOMA—much less common than vestibular schwannoma A. Tumor limited to the Meckel cave in the middle fossa—pterional craniotomy should provide sufficient exposure B. Dumbbell-shaped tumor with extension into the posterior fossa—more aggressive approach such as the transpetrosal C. Dissection may be epidural (find V2 and V3 and trace to the tumor) or intradural (Fig. 8.4). XV. HEMANGIOBLASTOMA—usually located in the posterior fossa near the sigmoid–transverse junction. A. Evaluation—evaluate for pheochromocytoma and renal cell carcinoma Fig. 8.4 Trigeminal schwannoma. (A) Axial and (B) coronal infused T1-weighted magnetic resonance images demonstrating a smooth, circumscribed, enhancing mass in the Meckel cave. (With permission from Citow JS. Neuropathology and Neuroradiology: A Review. New York, NY: Thieme Medical Publishers; 2001: 105, Fig. 148.) B. Treatment—surgical resection after embolization is performed similar to arteriovenous malformation (AVM) surgery by dissecting around the lesion and initially avoiding the veins. C. Only 20% are associated with Von Hippel-Lindau syndrome (CNS hemangioblastomas, retinal hemangiomas, renal cell carcinoma, pheochromocytoma, and cysts of the liver, pancreas, kidney). XVI. CHORDOMA—usually located in the sacrum or clivus A. Treatment—wide en bloc surgical resection and proton beam XRT; prognosis usually poor with median 10 year survival of 20–30% XVII. EPIDERMOID AND DERMOID TUMORS A. Treatment 1. Symptomatic—treat with surgical resection 2. Steroids—Mollaret meningitis (inflammation by leakage of cholesterol crystals) so avoid spillage during resection A. Evaluation 1. Hyperdense on computed tomography (CT), hyperintense on T1-weighted MRI, and enhances strongly like a meningioma 2. Examination of lymph nodes, chest x-ray (CXR), CT of the chest and abdomen, bone marrow biopsy, testicular ultrasound, and eye examination for uveitis B. Treatment—tumors may disappear with steroids. 1. Biopsy 2. XRT—50 Gy fractionated 3. Chemotherapy—intrathecal methotrexate in ventricular access device 12 mg twice a week × 6 with IV leucovorin rescue C. Survival—3 months without treatment, 10 months with XRT XIX. PAR AGANGLIOMAS A. Carotid body tumor 1. Signs/Symptoms—usually presents as a neck mass with CN X or XII palsy and ICA insufficiency 2. Evaluation—MRI and angiogram (external carotid artery feeders and a splayed bifurcation). The tumors may release catecholamines and 5% are bilateral. 3. Treatment—resection (frequent CN X and XII injury 40%, CVA 10%, and death 10%) and XRT B. Glomus jugulare or tympanicum 1. Signs/Symptoms—usually presents with CN IX–XII palsies (jugulare) or CN VII and VIII palsy and bruit (tympanicum). 2. Evaluation a. Evaluate with otoscope—pulsatile red/blue mass behind eardrum b. Urine vanillylmandelic acid (VMA)—tumors may release catecholamines, serotonin, or histamine c. MRI and CT 3. Treatment—surgical resection (with preoperative embolization) and XRT a. Histamine release at surgery may cause hypotension or bronchoconstriction; pretreat with 3 weeks of α-blockers and 3 days of β-blockers. XX. PEDIATRIC TUMORS—juvenile pilocytic astrocytoma, medulloblastoma, brainstem glioma, and ependymoma; in children < 1 year, usually teratoma and glioblastoma (GBM) XXI. SKULL TUMORS—epidermoid (scalloped margins), hemangioma (sunburst pattern), eosinophilic granuloma (punched-out lesion with clean margins, tender, associated with histiocytosis X), osteoma (sclerotic), and meningioma (hyperostotic) A. Treatment—en bloc resection if symptomatic 1. Consider radiating a hemangioma if patient unable to tolerate surgery B. Tumors originating from various tissues—bone (osteoma and osteosarcoma), cartilage (chondroma and chondrosarcoma), fibrous (meningioma), marrow (multiple myeloma and histiocytoma), vessel (hemangioma), and other (fibrous dysplasia, Paget disease, metastatic tumors) XXII. METASTASES A. Most frequent lesions—lung, breast, kidney, colon, and melanoma B. Tumors that frequently hemorrhage—choriocarcinoma, renal cell carcinoma, and melanoma C. Most common in children—neuroblastoma, rhabdomyosarcoma, and Wilms tumor D. Drop mets—possible from medulloblastoma, ependymoma, pineal tumors E. Evaluation—evaluate for primary tumor with CXR; CT of chest, abdomen, and pelvis; mammogram; bone scan 1. Areas of diffuse ependymal enhancement are seen with leptomeningeal carcinomatosis or abscess. F. Treatment 1. Anti-epileptic (e.g. keppra) and steroids; possibly surgical resection and XRT 2. Solitary met—excision and whole brain radiation therapy (WBRT) or stereotactic radiosurgery (SRS) if inaccessible. 3. 3 or less mets—WBRT and SRS or surgery if accessible with one craniotomy 4. Greater than 3 mets—WBRT 5. XRT—30 Gy over 2 weeks (sensitive tumors are small cell, germinoma, and lymphoma) 6. Postop XRT—questionable value G. Survival—consider resection only if greater than 6 months XXIII. NEUROFIBROMATOSIS TYPE I (NF-1) A. Inclusion criteria are at least two of the following: 1. Six café au lait spots 2. Two neurofibromas 3. One plexiform neurofibroma 4. Axillary or inguinal freckling 5. An osseous lesion (sphenoid dysplasia or thinning of long bones) 6. An optic glioma 7. Two Lisch nodules (iris hamartomas, only seen in NF-1) 8. A relative with NF-1 XXIV. NEUROFIBROMATOSIS TYPE II (NF-2) A. Associated tumors—bilateral vestibular schwannomas, meningiomas, astrocytomas, hamartomas, spinal ependymomas (spinal astrocytomas are more common in NF-1), and nerve root schwannomas B. Inclusion criteria—bilateral vestibular schwannomas or a relative with NF-2 and one vestibular schwannoma, or two of the following: 1. Neurofibroma, meningioma, glioma, schwannoma, or postcapsular cataract at a young age C. Treatment—attempt to remove the vestibular schwannomas when small and try to preserve hearing in at least one ear. Stereotactic radiosurgery may be of value. XXV. TUBEROUS SCLEROSIS A. Signs/Symptoms—Classical triad is adenoma sebaceum (rashlike facial angiofibromas), mental retardation, and seizures. B. Associated lesions 1. Cerebral hamartomas—tubers, frequently calcify, and usually don’t enhance 2. Subependymal giant cell astrocytoma—enhances and is located near the foramen of Monro 3. Cardiac rhabdomyoma 4. Renal angiomyolipoma 5. Pancreatic adenoma 6. Retinal hamartoma 7. Cysts in the lung, liver, and spleen C. Treatment—resect the astrocytoma if symptomatic (usually by hydrocephalus) XXVI. STURGE-WEBER SYNDROME A. Signs/Symptoms—ipsilateral V1 port-wine stain, cortical tram-track calcifications, and atrophy B. Treatment—anticonvulsants and a skin-colored tattoo on the nevus flammeus XXVII. RING-ENHANCING LESION—Vascular, Infectious, Neoplastic, Demyelinating (VIND) XXVIII. BASAL GANGLIA LESIONS—infection, astrocytoma, lymphoma, metastatic tumor, demyelination XXIX. TRANSDURAL NASAL MASS—juvenile angiofibroma, esthesioneuroblastoma (use chemotherapy before resection and XRT), mucormycosis, meningioma, and metastatic disease XXX. RADIATION A. Theory—x-rays and gamma rays deliver photons and particulate radiation to tumor cells to cause cell death and cessation of replication by freeing an electron to break bonds and produce free radicals to injure DNA. 1 Gy = 1 J of energy absorbed/kg 1 Gy = 100 rad 1 cGy = 1 rad B. External beam radiation 1. Fractionation divides the dose to increase the therapeutic ratio (effect on tumor cells/normal cells). 2. Results depend on dose, time, and field. The body repairs sublethal damage. 3. Three Rs are redistribution (cells are most susceptible in the mitotic phase), repopulation (cell types change), and reoxygenation (cells are more susceptible with increased oxygen). 4. Delay XRT until 10 days postsurgery to allow initiation of healing 5. Germinomas and lymphoma melt away but recur. C. Radiation necrosis 1. Vascular endothelium and oligodendrogliomas are the most susceptible. 2. Usually occurs after 18 months 3. Radiation therapy lowers IQ 25 points (especially doses > 40 Gy) and may injure the optic pathways (limited to 8 Gy) and the pituitary/ hypothalamic system, cause demyelination after methotrexate, and produce new tumors (GBM, meningioma, or osteosarcoma). 4. Evaluation—MR-spectroscopy or positron emission tomography (PET) scan (increased glucose use in tumors decreases with necrosis) 5. Treatment a. Only resect necrosis if there is mass effect b. Prevent necrosis by keeping the radiation dose < 60 Gy over 6 weeks; in the spinal cord avoid doses > 0.2 Gy/d D. Stereotactic radiosurgery 1. General a. Best for AVMs or tumors < 3 cm and inaccessible b. 2-year latency with AVM therapy before the hemorrhage risk decreases c. Use multiple ports with a steep gradient drop-off to decrease surrounding brain injury 2. Linear accelerator (LINAC) a. Photon beam is made by electron acceleration (x-ray). b. More flexible and less expensive than gamma knife with non-spherical lesions c. Results—similar to gamma knife d. Modification of target—produced by changing arc paths and collimator size 3. Gamma knife a. Photon beam is made by natural radioactive decay (gamma ray) b. Narrower distribution of energy produces slightly better spatial accuracy than LINAC, but this is less than the error in target margins (LINAC has 1 mm error). c. Uses different-sized collimators and exposure times and > 1 isocenter d. Target modification—performed by plugging collimators passing radiation through sensitive areas 4. Fractionated stereotactic radiosurgery (radiotherapy) a. Reoxygenation helps kill more cells. b. Fractionation hits late responders and multiple cell cycles. c. Hypofractionation—1 fraction/d × 1 week; best for slow-growing benign tumors (meningioma, schwannoma, and AVM) 5. Planning a. Use five arcs 100 degrees each to keep the drop-off optimal. b. Tissue 2.5 mm away will be injured. c. CT localizes better than MRI due to magnetic artifact, but accuracy can’t be < 0.6 mm due to pixel size (MRI shift is 2 mm). d. Multiple isocenters help conform to an irregular shape 6. Specific lesions a. AVM—perform 30 days after embolization (avoid radiopaque dye). Use 15 Gy to the periphery of the lesion. Obliteration rate is 50% at 1 year and 86% at 2 years. b. Schwannoma—use 14 Gy at 80% isodense line; 40% shrink and 40% stop growing c. Metastatic tumors—use 15 Gy at the center at 80% isodense curve 7. Complications—headache, emesis, seizures (premedicate with Deca -dron 10 mg and phenobarbital 90 mg), radiation necrosis 3%, symptomatic white matter changes 20%, vascular changes 5%, and CN dysfunction 1% Helpful Hints
8: BRAIN TUMORS
Only gold members can continue reading. Log In or Register to continue


Full access? Get Clinical Tree


