Acute Physiologic Changes, Morbidity, and Mortality of Status Epilepticus
Simon D. Shorvon
John M. Pellock
Robert J. DeLorenzo
Introduction
Status epilepticus is a relatively infrequent but serious manifestation of epilepsy. In population-based studies, annual incidence rates have been shown to vary between 9.9 and 41 per 100,000.18,25,26,27,28,29,30,31,32,44,49,51,56,85,88,99,106 The highest annual incidence rate (41 of 100,000) was found in the prospective population-based study carried out by De Lorenzo et al. in Richmond, Virginia (Fig. 1). The age distribution of the frequency of total status epilepticus events per year and the incidence of status epilepticus are presented in FIGURE 2. Extrapolating these data, 102,000 to 152,000 individuals would be affected each year in the United States (with 126,000 to 195,000 status epilepticus episodes), and over 22,000 to 42,000 deaths per year occurred in patients with status epilepticus. From hospital studies, Hauser44,45 estimated that status epilepticus occurs in 50,000 to 60,000 individuals in the United States annually: One third of these patients present with status epilepticus (SE) as a first unprovoked seizure; one third present with established epilepsy; and one third present with no previous history of epilepsy. Recent cost studies from the Richmond group have developed estimates of the cost of SE in an urban hospital.76 The estimate from this data indicated that the cost for care of SE in the United States per year is approximately $3 billion to $4 billion. Projecting the population of the United States to the world would suggest that SE would cost more than $70 billion to $93 billion per year on an international basis.
The differences in these incidences may reflect the different methods of data collection and the ability to correctly identify cases of SE from the medical record. Analysis from the Richmond study indicates that it is very difficult to identify cases of SE from International Classification of Diseases (ICD)-9 codes, discharge summaries, and retrospective chart reviews.26,27 The higher incidence in the Richmond study may in part be due to the prospective nature of this study and the ability of this study to detect nonconvulsive SE cases. Nonconvulsive SE is often not detected unless aggressive electroencephalographic (EEG) monitoring is conducted on all comatose patients. Nonconvulsive SE was found in 8% of comatose patients in the Richmond study.98 In addition, environmental, genetic, racial, and other differences may contribute to the ranges in incidences for SE in these studies. Race is a significant contributing factor in determining the incidence of SE.26,27 Studies from Richmond had an incidence of SE of 41 per 100,000.28 This incidence was about one to four times that of SE in the predominantly white (Caucasian) populations of other studies.18,44,45,49,51,56,85,88,99,106 The incidence of the white population in Richmond was 19 per 100,000,28 which compared well to several other predominantly white populations.44,45,51,56,88,106 However, the nonwhite, African American population in Richmond had a much higher incidence of SE, 57 per 100,000.28 Since Richmond is over 50% African American, the higher incidence in this group accounted for the higher incidence in the overall Richmond population. Correcting the Richmond data for the higher percentage of African Americans in the population gives an incidence very close to the Rochester44,45 and Hessen51 studies. Further investigations are needed to more fully evaluate the effects of genetic factors and race on the incidence of SE. It is essential to further evaluate these ethnic disparities in the incidence and mortality of SE around the world.
Status epilepticus can cause cerebral damage in various ways.30 The damage caused by physiologic changes that are a direct consequence of seizure activity (“excitotoxic” damage) is usually considered the most important mechanism of damage, although status epilepticus can cause other physiologic changes that also may result in cerebral damage. The drug therapy of status epilepticus also carries risks. The situation is complex and these variables are often interrelated, and skillful medical management is essential to minimize the physiologic changes, morbidity, and mortality. Most studies have been carried out in tonic–clonic status epilepticus, and it is clear that the risk of morbidity and mortality is highest in this form. In this chapter, only this form of status epilepticus will be considered.
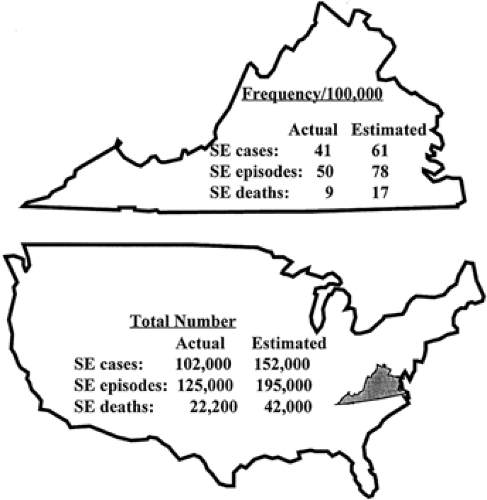 FIGURE 1. Schematic representation of the frequency and total number of status epilepticus (SE) episodes and status epilepticus deaths in Richmond, Virginia (top), and the United States (bottom). “SE cases” represents the number of patients presenting with one episode of status epilepticus in a year. “SE episodes” represents the number of occurrences of status epilepticus per year. “SE deaths” represents the number of deaths associated with status epilepticus per year. The data represent the actual and estimated values based on the validation of the case ascertainment studies in Richmond (see Materials and Methods). The estimates projected for the United States assume that Richmond, Virginia, is a representative population for the country. Further corrections for the distribution of factors such as age, race, and etiologic variations between Richmond and the United States are necessary to make more accurate projections. Studies are in progress at the VCU/MCV Comprehensive Epilepsy Institute to obtain more accurate estimates of the rates of occurrence and mortality for status epilepticus in the country and the world. (Reprinted with permission from Towne AR, Pellock JM, Ko D, et al. Determinants of mortality in status epilepticus: a retrospective study of 292 adult patients. Epilepsia. 1994;35:27–34.) |
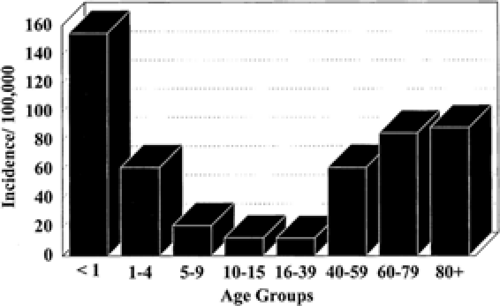 FIGURE 2. Age-specific distribution of the incidence of status epilepticus per year per 100,000 population in Richmond, Virginia. Incidence of status epilepticus represents the number of patients who developed a first episode of status epilepticus in Richmond per year per 100,000 population in each age group. These data do not include recurrent episodes of status epilepticus. The population for each age group was obtained from the 1990 U.S. Census Bureau for Richmond, Virginia. (Reprinted with permission from Towne AR, Pellock JM, Ko D, et al. Determinants of mortality in status epilepticus: a retrospective study of 292 adult patients. Epilepsia. 1994;35:27–34.) |
Systemic Physiologic Changes in Status Epilepticus
These have been reviewed in detail elsewhere.10,22,30,64,85 The majority of information comes from tonic–clonic SE, but some data exist from other types of SE. Below we list the changes that can influence outcome in tonic–clonic status epilepticus. Monitoring and skillful management of these changes are vital.
Blood Pressure and Heart Rate
Generalized seizures in animals and humans result in elevated systemic arterial pressures. Similar changes occur in the early phases of convulsive status epilepticus and levels occasionally reach those encountered in hypertensive encephalopathy. In the 21 patients of White et al.,103 whose blood pressures were recorded during pentylenetetrazol-induced seizures, the mean elevation in systolic pressure was 85 mm Hg and in diastolic pressure 42 mm Hg (maximum changes over baseline were 120 mm Hg for systolic and 100 mm Hg for diastolic, with the minimum changes being 25 and 10 mm Hg, respectively). The time course of seizure-induced hypertension has been studied more thoroughly in animals.65,90 In the setting of bicuculline-induced status epilepticus in sheep, systemic pressure, heart
rate, and plasma epinephrine/norepinephrine concentrations each peaked within the first minute of seizure activity (Fig. 3). Systemic pressure elevations of approximately 150% then returned gradually to baseline over the first 60 minutes of status epilepticus, whereas epinephrine and norepinephrine concentrations as well as heart rate showed only modest attenuation. Benowitz et al.8 speculated that the mechanism responsible for the decline in blood pressure despite persistent sympathetic activation during status epilepticus was desensitization of vascular adrenergic receptors or hypovolemia induced by vasoconstriction with the resultant movement of blood volume into extracellular spaces. Plasma catecholamine levels after spontaneous single seizures89 in humans are presented in FIGURE 4. With single seizures or a flurry of seizures, systemic hypertension occurs as well. Again, the peak pressures occur within the first minute, and the maximum level reached is not different between one seizure, a flurry of seizures, or status epilepticus7 (Fig. 5).
rate, and plasma epinephrine/norepinephrine concentrations each peaked within the first minute of seizure activity (Fig. 3). Systemic pressure elevations of approximately 150% then returned gradually to baseline over the first 60 minutes of status epilepticus, whereas epinephrine and norepinephrine concentrations as well as heart rate showed only modest attenuation. Benowitz et al.8 speculated that the mechanism responsible for the decline in blood pressure despite persistent sympathetic activation during status epilepticus was desensitization of vascular adrenergic receptors or hypovolemia induced by vasoconstriction with the resultant movement of blood volume into extracellular spaces. Plasma catecholamine levels after spontaneous single seizures89 in humans are presented in FIGURE 4. With single seizures or a flurry of seizures, systemic hypertension occurs as well. Again, the peak pressures occur within the first minute, and the maximum level reached is not different between one seizure, a flurry of seizures, or status epilepticus7 (Fig. 5).
With prolonged status epilepticus, blood pressure decreases to levels below baseline.8,63 The risk of cerebral hypoperfusion then arises due to the inhibition of cerebral autoregulation during seizures due to a decrease in cerebral vascular resistance.78 In experimental primates, boundary zone lesions occurred in the cortex and cerebellum when blood pressure was <75 mm Hg during the final 30 minutes of status epilepticus.65 Drug therapy can also greatly lower blood pressure, and this is a common clinical problem, for instance, with the use of bolus doses or prolonged infusion of anesthetic agents, barbiturates, phenytoin, and benzodiazepines. Pressor agents are often required during anesthesia in status epilepticus, and blood pressure must be carefully monitored.
Cardiac Effects
Status epilepticus can have significant effects on cardiac function. The Richmond study has provided direct evidence in humans of the effects of SE on cardiovascular function.11,12 These studies indicate that during or immediately after control of SE, patients exhibit a high frequency of electrocardiographic (ECG) abnormalities when compared with expected rates of abnormalities in control populations. Several ECG abnormalities, including ischemia, conduction defects, prolongations of the QTc interval, the presence of latent potentials on sinoatrial ECGs, and the absence of the expected sinus tachycardia during SE, correlated significantly with mortality.12 Monitoring patients after SE and up to the time of death with cardiac monitoring, it was found that at the time of death after SE, two distinct cardiovascular patterns of mean arterial pressure (MAP) and heart rate (HR) were observed.11 One group of patients manifested a gradual decline in MAP and/or HR (GDP), and the other group showed sudden death with an acute cardiac decompensation pattern (ADP). These studies represent the only large comprehensive, prospective, population-based data to evaluate central nervous system (CNS) and cardiac factors in the mortality of generalized tonic–clonic, partial complex, and nonconvulsive SE in humans.
Acidosis
A severe lactic acidosis is a prominent accompaniment of status epilepticus in humans and animals5,64,72 (Fig. 6). Arterial pH measurements in 70 spontaneously ventilating patients with status epilepticus ranged from 6.28 to 7.5; the pH was <7.35 in 59 patients and <7.0 in 23 patients.5 A respiratory component to the acidosis was found in 30 patients, of whom 13 had a recorded PCO2 over 60 mm Hg. The decrease in blood pH
is rapid (from 7.33 to 6.83 within 4 minutes in rats),91 reaching minimal levels within 15 to 20 minutes of status epilepticus in baboons (6.47 to 6.86).65 The rapid rate of decrease of pH is also demonstrable by the similar pH values found after a single seizure: 6.86 to 7.36.72 A respiratory component to the acidosis in a single seizure also was seen (PCO2 range 31 to 65 mm Hg). The pH then normalizes over 60 minutes as the causative agent, lactic acid, is metabolized. Other potential causes of acidosis include an acceleration of glycolysis, tissue hypoxia, and catecholamine release. Seizure-induced acidosis is markedly attenuated by treatment with neuromuscular paralysis,67,91 showing that anaerobic metabolism in muscle is the major source of lactate accumulation. In well-ventilated and paralyzed patients, acidosis is not usually a major clinical problem, and although the administration of bicarbonate can be helpful, this is not usually required and should not be given routinely.85
is rapid (from 7.33 to 6.83 within 4 minutes in rats),91 reaching minimal levels within 15 to 20 minutes of status epilepticus in baboons (6.47 to 6.86).65 The rapid rate of decrease of pH is also demonstrable by the similar pH values found after a single seizure: 6.86 to 7.36.72 A respiratory component to the acidosis in a single seizure also was seen (PCO2 range 31 to 65 mm Hg). The pH then normalizes over 60 minutes as the causative agent, lactic acid, is metabolized. Other potential causes of acidosis include an acceleration of glycolysis, tissue hypoxia, and catecholamine release. Seizure-induced acidosis is markedly attenuated by treatment with neuromuscular paralysis,67,91 showing that anaerobic metabolism in muscle is the major source of lactate accumulation. In well-ventilated and paralyzed patients, acidosis is not usually a major clinical problem, and although the administration of bicarbonate can be helpful, this is not usually required and should not be given routinely.85
 FIGURE 3. A: Responses of systemic arterial blood pressure and heart rate to status epilepticus induced by bicuculline injection (time 0). Seizure activity persisted for the duration of the experiment in paralyzed sheep. Data points represent mean (± standard error of the mean [SEM]) of five animals, except for heart rate at 120 and 180 minutes, which represent data collected from two animals. Asterisks indicate a significant difference from pressurized value (p <0.05). B: Plasma norepinephrine and epinephrine response to status epilepticus (mean ± SEM). Asterisks indicate a significant difference from pressurized value (p <0.05). (From Aminoff MJ, Simon RP. Status epilepticus. Causes, clinical features and consequences in 98 patients. Am J Med. 1980;69:657–666.) |
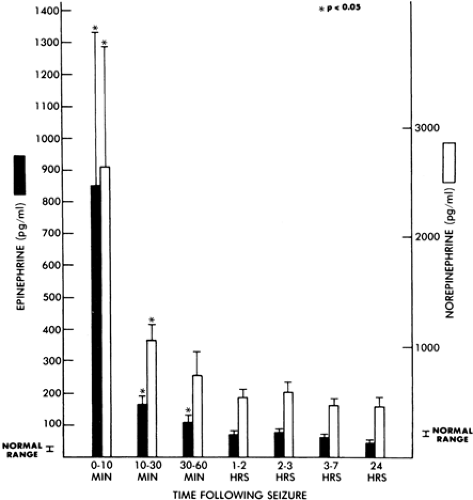 FIGURE 4. Plasma epinephrine and norepinephrine concentrations after a single generalized tonic–clonic convulsion. Data expressed as geometric means (± status epilepticus). (Reprinted with permission from Simon RP, Aminoff MJ, Benowitz NL. Changes in catecholamines after tonic clonic seizures. Neurology. 1984;34:255–257.) |
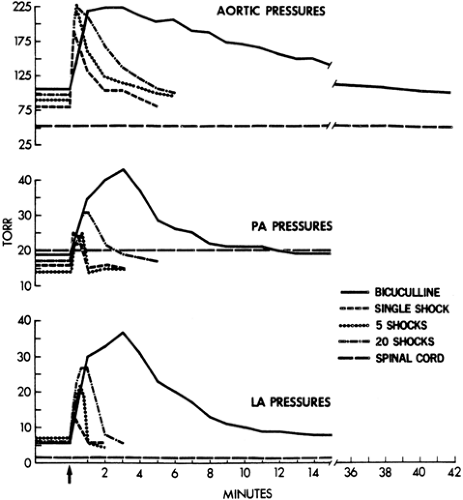 FIGURE 5. Vascular pressure elevations during seizures in sheep; mean values, plotted at 10-second intervals. “Spinal cord” refers to animals with cervical spinal cord transection before seizures. “Single shock,” “5 shocks,” and “20 shocks” refer to numbers of electroconvulsive seizures induced. “Bicuculline” refers to bicuculline-induced status epilepticus. PA, pulmonary arterial; LA, left atrial. (Reprinted with permission from Bayne LL, Simon RP. Systemic and pulmonary vascular pressures during generalized seizures in sheep. Ann Neurol. 1981;10:566–569.) |
The induced acidosis is not thought to be generally correlated with the degree of neuronal injury in status epilepticus. In the classic studies of Meldrum and Brierley,64 the mean arterial pH during the last 30 minutes of status epilepticus was 6.92, 7.08, and 7.14 in animals with no, moderate, or severe CNS damage, respectively. Using heat shock protein immunocytochemistry as a marker of neuronal injury in experimental status epilepticus, acidotic animals (paralyzed and ventilated with 10% CO2) had less neuronal injury than did animals with normal pH. However, the apparent protective effect was due to attenuation of seizure duration in the acidotic animals.79 It is also worth noting that acidosis has a strong anticonvulsant effect (a fact underpinning the use of the ketogenic diet in epilepsy, for instance).104
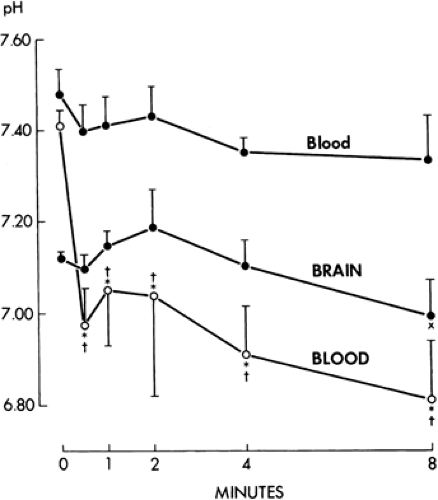 FIGURE 6. Blood and brain pH during status epilepticus. •, paralyzed–ventilated animals; ○, nonparalyzed (freely convulsing) animals (brain pH not determined in freely convulsing animals). Status begins at time 0. Data plotted as means ± standard deviation. *p <0.001 control value (time 0) versus each time point for blood pH in freely convulsing seizures. p <0.001 blood pH paralyzed versus nonparalyzed animals at each time point. ×p <0.005 brain pH, time 0 versus all time points. (Reprinted with permission from Simon RP, Aminoff MJ, Benowitz NL. Changes in catecholamines after tonic clonic seizures. Neurology. 1984;34:255–257.) |
Hypoxia
Arterial oxygen tension decreases modestly in status epilepticus.64,65,80 The mean arterial PO2 values were 58 to 68 mm Hg in experimental primates that showed no moderate or severe neuropathic change.64 Results of human and experimental studies support the concept that hypoxia alone does not induce brain damage, at least in the early stages of status epilepticus.13,42 In fact, there is some evidence that hypoxia may be neuroprotective during seizures. Blennow et al.10 demonstrated that a reduction of arterial PO2 to 50 mm Hg during the 2 hours of experimental status epilepticus protected animals from neuropathic damage in the cortex hippocampus and striatum; prominent ischemic cell change occurred in animals with induced status epilepticus without hypoxia. Blennow suggested that its protection was due to an attenuation of seizure intensity as measured electroencephalographically. Amano et al.4 also noted the ameliorative effect of hypoxia (8.5% oxygen with balanced nitrogen) upon seizure induction and excitotoxic injury resulting from intravenous kainic acid. On the other hand, maximal oxygenation (ventilation with 100% oxygen) does not exacerbate neuropathic change in experimental status epilepticus.94 As status epilepticus proceeds, however, it is likely that prolonged hypoxia does carry a risk of serious damage via many potential mechanisms including free radical release, rupture of lysosomes, and the induction of intracellular acidosis and apoptotic cascades. The increased demands for oxygen are not confined to cerebral tissue, and there are impressive metabolic demands from convulsing muscles. The protection of respiratory function and administration of oxygen is an important element of the emergency management of convulsive status epilepticus.
Pulmonary Edema
Status epilepticus affects not only inspiratory and expiratory effort, but also the passage of fluid across the pulmonary capillary bed. The barrier to this movement is at the pulmonary capillary. The major clinical problem is the occurrence of pulmonary edema. This was found in 15 of 41 patients who died during status epilepticus71 and in eight patients with the diagnosis of epileptic sudden death.95 Repetitive seizures or status epilepticus are more likely to be followed by pulmonary edema than by a single seizure.20 The best experimental studies have been carried out in sheep, and status epilepticus resulted in increases in pulmonary arterial pressure of up to 685% of that in resting states.7 The greater the number or the longer the duration of seizures, the higher are the increases in intravascular pressures (Fig. 6), and the fall in pulmonary arterial pressure after seizures lags behind that of the systemic arterial pressure. These pulmonary arterial pressures are well in excess of the osmotic pressure of blood, and the lung capillaries also suffer from stretch injury. These marked increases in intravascular pressure in the pulmonary capillaries cause a doubling of pulmonary lymphatic drainage, an increase in transcapillary albumin conductance, and an alteration in pulmonary capillary permeability90 (Fig. 7).
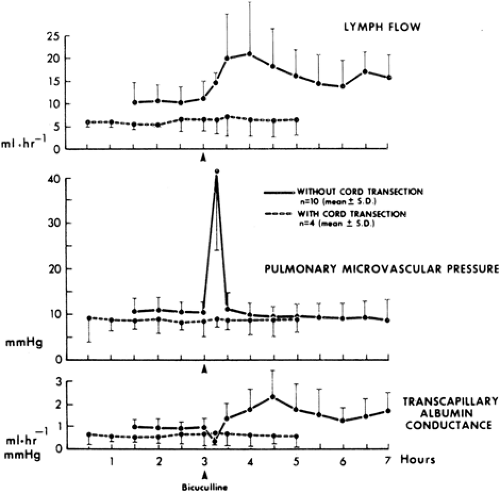 FIGURE 7. Pulmonary lymph flow, calculated pulmonary microvascular pressure, and calculated pulmonary transcapillary albumin conductance before and during bicuculline-induced status epilepticus in ten paralyzed, halothane-anesthetized sheep (solid line) and four sheep with cervical spinal cord transection (broken line). Values are presented as means ± standard deviation. Arrowheads indicate administration of bicuculline. (Reprinted with permission from Simon RP, Bayne LL, Tranbaugh RF, et al. Elevated pulmonary lymph flow and protein content during status epilepticus in sheep. J Appl Physiol. 1982;52:91–95.) |
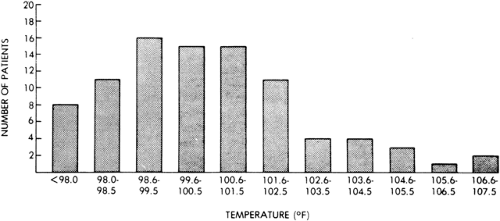 FIGURE 8. Rectal temperatures recorded between seizures or immediately after institution of treatment in 90 patients with status epilepticus. (Reprinted with permission from Aminoff MJ, Simon RP. Status epilepticus. Causes, clinical features and consequences in 98 patients. Am J Med. 1980;69:657–666.) |
Body Temperature
Hyperthermia is a very common accompaniment of major motor status epilepticus. The convulsing muscles and the sympathetic overactivity due to the heat produce the rise in temperature. The importance of the latter is shown by the increase in body temperature in paralyzed animals.100 In baboons paralyzed at the onset of status epilepticus, a mean temperature
increase was 2.05°C (range 1.0 to 2.7) over a 7-hour period of observation. In freely convulsing experimental primates, a core temperature of >40°C, persisting for more than 3 hours, was associated with neuronal damage, greater than that predicted from the seizure duration alone.64,100 The neuronal damage was most prominent in the cerebellum. In these animals, the temperature elevation of 40°C was reached within 60 to 90 minutes of status epilepticus onset.100
increase was 2.05°C (range 1.0 to 2.7) over a 7-hour period of observation. In freely convulsing experimental primates, a core temperature of >40°C, persisting for more than 3 hours, was associated with neuronal damage, greater than that predicted from the seizure duration alone.64,100 The neuronal damage was most prominent in the cerebellum. In these animals, the temperature elevation of 40°C was reached within 60 to 90 minutes of status epilepticus onset.100
In 90 patients in status epilepticus, hyperthermia was found in 75, with core temperatures reaching 107°F (42°C) (Fig. 8). The highest temperatures were found in patients with prolonged status epilepticus (more than 9 hours) and were
associated with brain injury.5 The duration of hyperthermia persisting after status epilepticus cessation can be estimated from reports of temperature elevation after single seizures. Most patients remained febrile at 12 hours, but only 3 of 27 were febrile at 48 hours.
associated with brain injury.5 The duration of hyperthermia persisting after status epilepticus cessation can be estimated from reports of temperature elevation after single seizures. Most patients remained febrile at 12 hours, but only 3 of 27 were febrile at 48 hours.
Leukocytosis
An elevation in the peripheral white blood cell count in status epilepticus is common, possibly due to the rapid elevation in plasma catecholamine concentrations. Such demarginated leukocytes resulted in white blood cell counts of 12,700 to 28,800 cells/mm3 in 50 of 80 patients in status epilepticus in whom there was no evidence of infection.5 A polymorphonuclear predominance was found in 17 of these patients, a lymphocytic predominance in 11, and a differential cell count with a normal distribution in 15.
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


