(1)
Asheville, NC, USA
The search for a specific biochemical cause of Alzheimer’s disease began in the 1960s and 1970s, and was inspired by the burgeoning success of levodopa treatment for Parkinson’s disease. In the case of Alzheimer’s disease, brain regions associated with higher brain functions, primarily the hippocampus and neocortex, are affected. Investigations of those regions in the presence of senile dementia led to the discovery that (1) there are deficits in presynaptic terminals in the enzyme that catalyzes the synthesis of the neurotransmitter acetylcholine (ACh), namely, choline acetyltransferase (ChAT); (2) acetylcholine has a role in learning and memory, and (3) blocking its release leads to memory impairment.
This collective set of findings gave rise to the cholinergic hypothesis of Alzheimer’s disease. That hypothesis was articulated in an influential 1982 paper by Bartus and led to the development of a number of drugs that act as cholinesterase inhibitors, that is, as agents that prevent acetylcholinesterase (AChE) from hydrolyzing acetylcholine in the synaptic cleft. Since that time it has become increasingly apparent that the primary effects of cholinergic depletion are on attention and only small improvements in symptoms of AD are seen in patients receiving e ACh-directed drugs. Following the discoveries of the involvement of amyloid-β in AD the main focus shifted away from the early groundbreaking observations on the cholinergic system to the glutamatergic system, the other main central excitatory neurotransmitter, and to pathologies derived from misfolded amyloid β.
8.1 The Amyloid Cascade Hypothesis
By the early 1990s the amyloid cascade hypothesis had replaced the cholinergic hypothesis as the leading idea as to the cause of Alzheimer’s disease. This articulation has been revised and expanded since its initial formulation in 1992 by Hardy and Higgins. In its initial form, the amyloid cascade hypothesis had as its main focus the buildup of amyloid plaque s that collect in the extracellular spaces. These deposits were thought to initiate a cascade of events leading to the development of the neurofibrillary tangle s and the fatal loss of neurons in the affected regions of the brain resulting in dementia and death.
Several key developments guided and supported this hypothesis. The first of these, as discussed in Chap. 1, was the identification in 1984 by Glenner and Wong of the 39–43 amino acid long amyloid β peptides as the main component of the extracellular plaque s seen in Alzheimer’s disease. That finding was followed by Masters’ confirmation in 1985, and by the discovery in 1987 of the gene that encodes the amyloid precursor protein on chromosome 21. The discovery was followed by that of the presenilin genes, PSEN1 and PSEN2, on chromosome 14 and 1, respectively, that encode proteins involved in processing the amyloid precursor protein, further strengthened the case for Aβ. In these genetic studies, the focus was on individuals suffering from early onset (ages less than 60 years) autosomal dominant familial forms of the disease (EOFAD, or EOAD). It is customary to distinguish between the early familial forms and later sporadic and familial forms of Alzheimer’s disease, or LOAD. In the latter case, the main risk factor is the presence of the apolipoprotein E4 (ApoE4) allele. The ApoE protein has a role in Aβ clearance, thereby supplying yet another connection between Aβ and Alzheimer’s disease.
However, it was soon noted that the correlations between the presence and severity of the extracellular amyloid plaque s and the clinical progression of the illness were weak, at best. The amyloid cascade hypothesis was then modified to reflect these findings along with the growing belief that the main toxic species was not the large and highly visible fibrillar deposits, but rather the smaller, more mobile and highly reactive Aβ oligomers. That shift in focus was promoted by a convergence of results of studies of several different neurodegenerative disorders including AD. The revised and current cascade hypothesis has as its main theme an inappropriate buildup of oligomeric species as the key initiator of dysfunction. That leaves as the paramount challenge identifying the exact manner or manners in which the Aβ oligomers are toxic with the most likely sites being the synapses and the blood–brain barrier, leading to progressive loss of neurons and failing neural circuitry.
This chapter presents the amyloid cascade hypothesis of Alzheimer’s disease as it currently stands. It begins with an examination of the two types of sequential processing of the amyloid precursor protein by proteolytic enzymes, and with an overview its possible regulation by trafficking, transcription, translation, and epigenetic elements. This is followed by a brief introduction to the different oligomeric species produced when there is an imbalance between production and clearance. This issue is particularly relevant given that a unique toxic species has not been identified and may not exist. This introduction is then followed by an overview of the three main clearance mechanisms. As already noted, the single most important genetic risk factor for developing LOAD is the presence of the ApoE4 allele. How expression of this factor influences Aβ aggregation and toxicity is not yet apparent but there are a number of plausible scenarios each supported by a growing body of data. These are explored in the content of Aβ clearance.
The blood–brain barrier and neurovascular unit are examined next. During the last few years the blood–brain barrier and the associated neurovascular unit have drawn increasing attention. Alzheimer’s disease is a multifaceted disorder. The “old” idea of a vascular dementia has been reborn within a broader framework whereby neural and vascular damage coexist and impact one another. A central tenet of the amyloid cascade hypothesis is that of synaptic damage. Synaptic dysfunction is explored along with findings that many of the key molecular players are lipid raft residents. Those discussions are accompanied by overviews of how inflammation , vascular dysfunction, and the cholinergic system impact the disorder, and then the second key observation of Alzheimer—the presence of neurofibrillary tangle s —is examined. Tau is the principal component of the neurofibrillary tangles, and is examined in the context of how it may act as a downstream effector of Aβ toxicity. Its broader set of actions awaits the chapter on tauopathies .
8.2 APP Processing and Generation of the Amyloid-β Protein
The Aβ peptide is derived from a larger β-amyloid precursor protein (APP) through a series of proteolytic cleavage operations. These are carried out by enzymatic proteins and protein complexes referred to as α-secretases , β-secretases, and γ-secretases. The presenilins, key risk factor s in EOAD, are part of the γ-secretase complex, which has three other members—Nicastrin, Aph-1, and Pen-2. As illustrated in Fig. 8.1 the presenilins are 8-pass transmembrane proteins proteolytically cleaved into two chains. A pair of aspartate residues positioned in opposition to one another is crucial for presenilin’s APP cleavage actions. APP passes through the gap formed by the segments containing the aspartates, and is cleaved. The presenilins carry out their catalytic activities together with the single-pass Nicrastrin protein, the 7-pass Aph-1 protein, and the 2-pass Pen-2 protein, which help assemble and stabilize the complex.
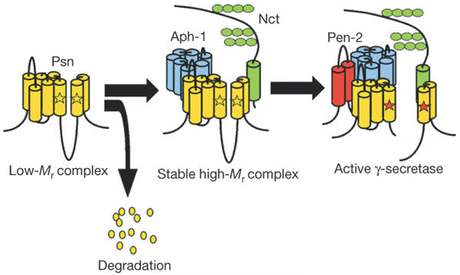
Fig. 8.1
Model of assembly and activation of the catalytically active γ-secretase complex. Psn Presenilin, Aph-1 anterior pharynx defective-1, Nct Nicastrin, Pen-2 presenilin enhancer-2. The positions of the critical aspartate residues are indicated by the pair of opposed stars (from Takasugi Nature 422: 438 © 2003 Reprinted by permission from Macmillan Publishers Ltd)
8.2.1 APP Processing
The processing of type I single pass transmembrane APP by the secretases involves making two cuts to the protein—the first cut is made outside the membrane and the second one within the lipid bilayer. The second type of processing is a fairly recent discovery and is known as regulated intramembrane proteolysis (RIP) (see Appendix 1). In the case of APP, there are two distinct ways of making the two cuts. The first way, the non-amyloidogenic pathway, uses the α-secretase to make the first cut. The second way, the amyloidogenic pathway, does not utilize the α-secretase but instead employs the β-secretase and that cut is made in a different location. The γ-secretase and its presenilins carry out the second cut in both routes.
In more detail, APP is cleaved at one of two alternative extracellular locations termed the α- and β-sites. This operation is commonly referred to as ectodomain shedding because an ectodomain stub is shed into the luminal space. The γ-secretases that carry out the subsequent cleavages create two fragments—an APP intracellular domain (AICD) and either the P3 or Aβ peptide depending on which enzyme carries out the first cut. As illustrated in the upper part of Fig. 8.2 the ectodomain generated by the α-secretase is designated as APPsα (or alternatively as sAPPα). In the second step, the γ-secretase generates the AICD and a P83 fragment. These operations may be contrasted with the amyloidogenic processing by the β- and γ-secretases shown in the lower portion of Fig. 8.2. In this situation, an sAPPβ stub is created and in the following cut the AICD and Aβ peptide are produced. In the case of cleavage at β sites, but not the α-sites, the fragments include the Alzheimer’s disease-causing 39–43-amino-acid-residue forms of the Aβ amyloid protein. The 42 residue form is favored by the mutated Presenilins, and is especially prone to aggregation.
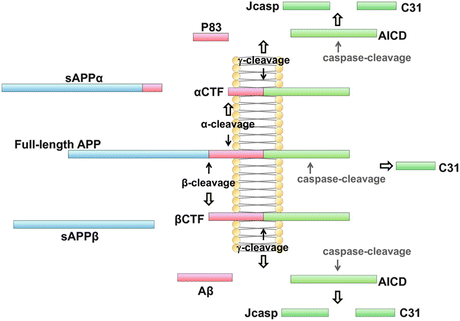
Fig. 8.2
Nonamyloidogenic and amyloidogenic processing of the amyloid precursor protein by α-, β-, and γ-secretases . Full-length APP undergoes non-amyloidogenic cleavage by the α- and γ-secretases, and amyloidogenic cleavage by the β- and γ-secretases (from Zhang 2011 Mol. Brain 4: art. 3; reprinted with permission through a Creative Commons Attribution License)
8.2.2 The Aβ Peptide
Aβ is a metalloprotein and, not surprisingly, elevated levels of Fe, Cu and Zn are present in amyloid plaque s . Increased amounts of these metals within the synaptic environment contribute to increases in oxidative stress and in the rates of aggregation and fibril growth. Support for these observations comes from the following. First, Aβ has high affinity binding sites for Cu and Zn, and Fenton chemistry is facilitated by these binding events resulting in an increased production of hydroxyl radicals. Secondly, α-secretases require Zn, β-secretase interacts with Cu and, in those situations where there is a lack of this metal, APP processing is affected. Thirdly, the presenilins facilitate Cu and Zn uptake and turnover. Lastly, the aggregation process itself is accelerated when Aβ coordinates Fe, Cu, or Zn.
Multiple isoforms of Aβ exist. Some of the isoforms are generated by proteolytic cleavage of the most N-terminal residues, while others are subject to a variety of posttranslational modifications. One particularly noteworthy modification is to the glutamine residue at position 3 (by glutaminyl cyclase) to form pyroglutamate, pE3. Modifications such as isomerization, metal-induced oxidation and phosphorylation, especially those involving the N-terminal-most residues, can accelerate the formation of aggregates and fibrils. The most prominent isoforms in the hippocampus and cortex of AD sufferers as determined by means of mass spectroscopy appear to be:
Aβ1–42
AβpE3–42
Aβ4–42
Aβ1–40
This bullet list is an ordered one; that is, the Aβ x–42 isoforms are thought to be more toxic than the Aβ x–40 ones. The reason for this difference is easy to understand upon noting that residues 41 and 42 are isoleucine and alanine, respectively (Fig. 8.3). Both of these residues are hydrophobic (Table 2.1) and their presence renders the peptide far more likely to form aggregates. In addition to these isoforms, a number of proteolytic enzymes may be active resulting in isoforms with x values ranging up to 10, Aβ1–y isoforms may be present, with y assuming values from 37 to 43, and various combinations of the two types of action may occur. The most prominent of the resulting isoforms are included in the bullet list.
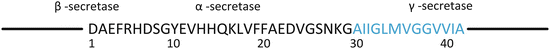
Fig. 8.3
The amyloid-beta (Aβ) peptide. The transmembrane portion of Aβ is shown in blue. The locations of the three secretase cleavage sites are shown above the sequence
8.2.3 Identity of the α- and β-Secretases
The α-secretases and β-secretases responsible for cleaving the APPs at the α- and β-sites are members of two families of proteases. One or more members of the “a disintegrin and metalloprotease” (ADAM) family function as α-secretases. The ADAM proteases are single pass transmembrane proteins with a N-terminal signal peptide, followed by a pro-domain. Two members of this family have roles in APP metabolism—ADAM17, also called TACE and ADAM10. ADAM10 has been identified as the main family members with ADAM17 perhaps acting as a backup.
The β-site APP cleavage enzyme 1 (BACE1) protein is the only beta secretase found to date. This transmembrane protein is a membrane-associated aspartyl protease belonging to the pepsin family. It possesses pre and pro domains followed by a catalytic domain, transmembrane helix, and cytoplasmic segment. BACE1 is the rate limiting enzyme in the formation of Aβ peptides .
8.3 Regulation of APP and Secretase Expression and Activity Occurs at Multiple Levels
APP and the secretases are tightly regulated at the trafficking, transcription, translation, and posttranslational levels. Full length APP is synthesized in the endoplasmic reticulum (ER). It is then transported through the trans-Golgi network (TGN), one of the main sites of APP residency in neurons. There it undergoes posttranslational modifications such as glycosylation and phosphorylation. It is transported to the cell surface in TGN-produced secretory vesicles where it may be cleaved by the α-secretases or reenter the endosomal pathway enclosed in clathrin-coated vesicles. Then, it may be sent back to the plasma membrane or transit to lysosomes for degradation. These trafficking steps are sketched in Fig. 8.4.
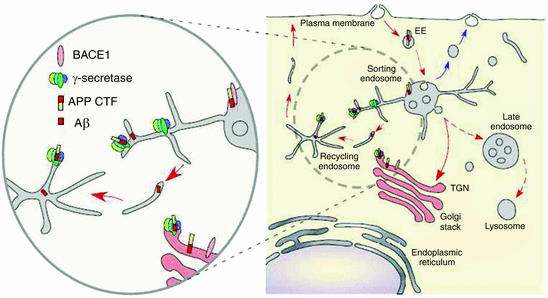
Fig. 8.4
Cellular trafficking of APP, Aβ, and BACE1. [The full-length protein (depicted as short bars) is synthesized in the ER, passes through the TGN to the cell surface (1) where is subject to recycling through the endosomal network (2) either back to the plasma membrane (3) or to the lysosomal compartment for degradation] (from Vetrivel Biochim. Biophys. Acta 1801: 860 © 2010 Reprinted by permission from Elsevier)
The β-secretase BACE1 follows a similar trafficking pattern, and in the Golgi its pro-domain is removed. It too cycles between the endosomes and plasma membrane. Its dwelling in the early endosomes permits it to come into contact with APP and cleave it, while residency in late endosomes leads to degradation in lysosomes. The four components that comprise the γ-secretase are synthesized and assembled in the ER. They then cycle between the ER and Golgi with a small number exiting to the plasma membrane and endosomal vesicles where they are able to carry out the second, intramembrane cleavage of the α- or β-CTF. The TGN and early endosomes function as sorting hubs where proteins are placed into cargo vesicles and sent to various locations. These processes are regulated. GGAs (Golgi-localized γ-ear-containing ARF binding proteins) mediate the trafficking between TGN and endosomes. Other complexes known as retromers regulate cargo selection and budding activities. All of these activities can influence the interactions between BACE1 and APP.
The promoter for BACE1 is a complex one with putative binding sites for a number of transcription factors. Its promoter is TATA-less with high CG content and with a Sp1 binding element (as is the case for APP and PrP), characteristics of a housekeeping gene. Other prominent binding sites are present, as well, such as those for NF-κB and PPARγ. The former regulates APP, β-secretase , and γ-secretase gene transcription. As has been found in many settings, the actions of NF-κB can be either repressive or stimulatory depending on the presence or absence of cellular stresses. Under healthy, non-stressed conditions NF-κB represses transcription of BACE1 but when the cells are continually exposed to stressful conditions such as those present in AD (e.g., high Aβ levels) NF-κB promotes BACE1 gene expression. PPARγ activity is thought to repress BACE1. However, activated microglia and astrocytes secrete pro-inflammatory cytokines and these signaling molecules suppress PPARγ leading to an increased production of BACE1. Overall, a positive feedback loop may well occur in which a resulting increased Aβ plaque buildup stimulates further inflammatory response leading to a greater production of BACE1 and hence more Aβ plaque buildup.
The expression levels of BACE1 are also regulated translationally and posttranslationally. One of the properties of the AD brain (and also of the non-AD brains of young/middle-aged individuals carrying the ApoE4 allele) is reduced glucose utilization. A link between reduced glucose utilization and BACE1 activity is provided by the translation initiation factor eIF2α. This control element increases BACE1, Aβ and APP levels when it is phosphorylated in response to energy deprivation stress. A second route whereby cellular stresses contribute to BACE1 activation is through actions by noncoding RNAs, most notably by the microRNAs miR-29a/b-1 and miR-107. MicroRNAs act as negative regulators of translation of substrate mRNAs. In AD brains, the activities of these microRNAs are reduced leading to an increased translation of BACE1 and a corresponding increase in Aβ levels.
8.4 Apolipoprotein E4 and Cholesterol Transport
Cholesterol is essential for proper synaptic and neuronal function. About 25 % of the total cholesterol in the body resides in the brain. Most of it resides in myelin sheaths with the remainder embedded in the plasma membranes of neurons and astrocytes. In short, cholesterol is:
A major component of the myelin sheath and needed for axonal growth and repair.
Used in synapse formation and remodeling, actions essential for learning and memory.
The key ingredient in lipid raft s , critical for synaptic transmission.
Cholesterol was first discovered in gallstones by François Poulletiere de la Salle in 1769. It was subsequently rediscovered and named cholesterine by Eugene Chevreul in 1815, and identified in blood by Boudet in 1833. As noted by Brown and Goldstein in their Nobel lecture, cholesterol has fascinated scientists ever since its discovery with 13 Nobel prizes having been awarded to scientists who studied this small molecule. Cholesterol cannot circulate by itself. Instead, it has to be transported by lipid transport vesicles—the lipoproteins (see Appendix 2). Uncovering the nature of this fat transport system has spanned three centuries—from its initial surmise by Robert Boyle in 1665 to the discovery of the low-density lipoprotein (LDL) receptor by Brown and Goldstein in 1974. Since then this transport system has been linked to a number of disorders, the most prominent being atherosclerosis and Alzheimer’s disease.
Apolipoproteins are amphipathic, lipid-binding protein constituents of the lipoprotein particles. There are several different kinds of apolipoproteins . These are designated by letters—A, B, C, etc. and sometimes numerically to distinguish different isoforms. For example, Apo B has two main isoforms, designed as B-48 and B-100. The former, a truncated version of Apo B-100, is synthesized in the gut and the latter is produced in the liver.
There are several different classes of lipoprotein particles. These, the lipids that they chaperone, and the responsible apolipoproteins are listed in Table 8.2 in Appendix 2. The apolipoproteins are large, and they wrap around and enable the transport of the hydrophobic lipids from one place to another in the body. For example, a typical LDL particle would contain one large apolipoprotein B particle plus about 2500 lipid molecules. These would be arranged in the following way. There would be core consisting of cholesteryl esters and triglycerides; plus an outer layer consisting of a smaller number of free cholesterol molecules plus a phospholipid monolayer. The entire assembly would be enveloped by the large apoB protein, which makes a pocket for the lipid ball.
Apolipoprotein E has three common isoforms, encoded by the APOE gene E2, E3, and E4 alleles. These differ from one another through the presence of either Cys or Arg at amino acid positions 112 and 158. The frequency of each of these alleles in the general population and among AD patients is presented in Table 8.1. As shown in the table the E3 allele is the most common and the E2 allele the least common. Possession of the E4 allele is the major risk factor for development of late onset familial and sporadic Alzheimer’s disease. Individual heterozygous for this allele have a 47 % chance of developing AD with a mean age of onset of 76 years. Individuals homozygous for this gene have an even greater chance of developing AD. For them the average age of onset decreases to 68 years with a frequency of 91 %.
Table 8.1
Apolipoprotein E2, E3, and E4 alleles and their frequencies in the general population and among AD patients (modified from Liu 2013 Nat. Rev. Neurol. 9: 106)
ApoE allele | Position 112 | Position 158 | Freq. (%) General | Freq. (%) AD |
|---|---|---|---|---|
E2 | Cys | Cys | 8 | 4 |
E3 | Cys | Arg | 78 | 59 |
E4 | Arg | Arg | 14 | 37 |
Apolipoprotein E (ApoE) is the main lipoprotein in the brain and cerebrospinal fluid (CSF). The abundance of ApoE in the brain is high with levels second only to that of the liver. The liver cannot supply the brain with ApoE since these particles are too large to pass through the blood–brain barrier (BBB ). Instead, the main suppliers of ApoE are the astrocytes with microglia and neurons providing the remainder. These cells maintain cholesterol and lipid homeostasis in the brain and ensure that the essential functions performed by cholesterol described above can be carried out.
8.5 Aβ Clearance and ApoE
Amyloid-β generation and clearance are complementary actions. Under healthy conditions, clearance is sufficiently robust and there is a balance between the two that prevents large deposits of amyloid-β from developing. Clearance of amyloid-β occurs in one of three ways (Fig. 8.5):
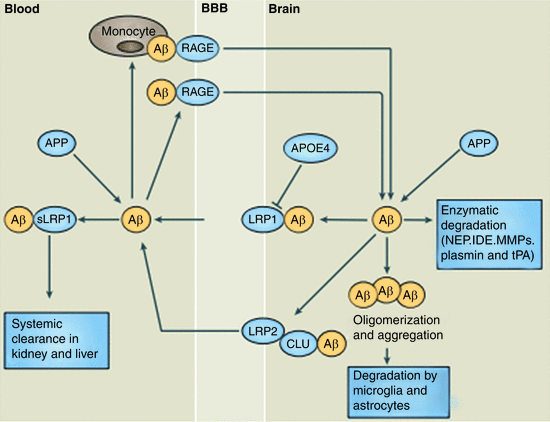
Fig. 8.5
Three methods of clearance of Aβ oligomers. LRP1, 2 low-density lipoprotein receptor-related protein 1, 2, CLU clusterin (also known as apolipoprotein J or ApoJ), RAGE receptor for advanced glycation end-products. See text for discussion (from Zlokovic Nat. Rev. Neurosci. 12: 723 © 2011 Reprinted by permission from Macmillan Publishers Ltd)
By means of enzymatic degradation;
Through uptake and lysosomal degradation by microglia and astrocytes, and
From export through the blood–brain barrier and clearance by liver and kidneys.
Several enzymes, most notably neprilysin (NEP), insulin-degrading enzyme (IDE), tissue plasminogen activator (tPA), and matrix metalloproteinases (MMPs), are responsible for degrading Aβ peptides . These enzymes reside in several places within multiple cell types and in the extracellular spaces either free or attached to the outer surface of the plasma membranes of those cells. Misfolded Aβ1–40 and especially Aβ1–42 resist degradation by aggregating, and this activity is enhanced by associations with heavy metals such as zinc and copper. These cations coordinate pairs of Aβ peptides, thereby facilitating aggregation into protease-resistant amyloid plaque s .
Astrocytes and microglia accumulate in senile plaques. They not only secrete Aβ-peptide degrading enzymes but also express receptors that enable their uptake and phagocytosis. Prominent among the receptors are members of the low-density lipoprotein receptor-related protein receptor (LRP) family, scavenger receptors, toll-like receptors (TLRs) and other danger receptors, complement receptors, and the receptor for advanced glycation end-products (RAGE). Aβ oligomers are difficult to deal with especially when present at high concentrations. As a result, phagocytosis is frequently impaired and an inflammatory response induced by the now activated glial cells. These cells release a variety of pro-inflammatory enzymes and reactive oxygen and nitrogen species. Interestingly, ApoE and especially the E4 allele compete with Aβ for binding to LRP1 and by that means ApoE can influence Aβ clearance.
APP is expressed throughout the body. Import of Aβ into the brain and export from the brain are mediated by RAGE and LRP1/2, respectively. RAGE is a multi-ligand receptor belonging to the Ig (immunoglobulin) superfamily. It is responsible for transporting unbound circulating Aβ peptides across the blood–brain barrier. Under normal conditions clearance by LRP is far more rapid than influx by RAGE, and the concentration of amyloid-β peptides in the brain is kept at a low level.
8.6 A Variety of Aβ Oligomeric Assemblages Are Formed
The revised and expanded amyloid cascade hypothesis posits that an inappropriate buildup of Aβ oligomers is the key initiating event in AD. Multiple lines of evidence acquired starting in the 1990s support this belief. In particular, the accretion of amyloid fibrils was found to correlate poorly with cognitive impairment, whereas the buildup of soluble, non-fibrillar Aβ oligomers correlated far more strongly with the severity of the disease. The production and secretion of soluble oligomers is a normal cellular process and thus by itself is not an indication of anything amiss. Rather, it is the production of large quantities of non-normal oligomeric assemblages that are the putative causative agents. These arise as a result of missense mutations in the APP proteins and presenilins, as a consequence of the ApoE4 allele, and due to gradually emerging age and stress-related factors including those associated with the metabolic syndrome.
A variety of oligomeric species may be formed from Aβ1–40 and the highly toxic Aβ1–42 peptides. Each of these species may populate several conformations endowed with specific set of toxicities, and these can interconvert from one oligomeric form to another. This multiplicity of assemblages and conformations creates difficulties in designing drugs aimed at countering their toxic effects. This aspect is illustrated in Fig. 8.6 in which the various oligomeric species that have been discovered are placed in two assembly pathways. One of these leads from monomers through trimers and through a set of non-fibrillar oligomers terminating in annular protofibrils. These structures bear a resemblance to bacterial toxins and for that reason it has been suggested that the Aβ oligomers are toxic through their ability to form pores in membranes. The other pathway has dimers as its basic building block and terminates with formation of amyloid plaques.
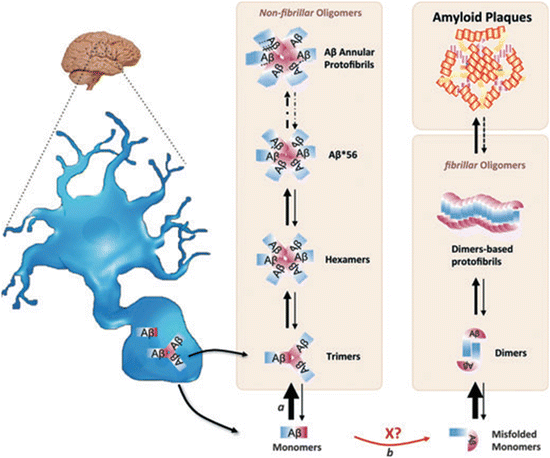
Fig. 8.6
Hypothetical dual pathway model of Aβ-oligomer assembly. Neurons secrete Aβ monomers and trimers. These can assemble and interconvert into larger-n species along the non-fibrillar pathway. The monomers may misfold, stimulated by unknown factor X, and form dimers. The dimers can assemble into higher-n oligomers and into the large fibrillar structures, the amyloid plaque s (from Larson J. Neurochem. 120: 125 © 2012 John Wiley and Sons, and reprinted with their permission)
Soluble and stable oligomers of various sizes and morphologies have been designated in several ways. One of these is as Aβ-derived diffusible ligands, or ADDLs. This term is intended to highlight their role as ligands for synaptic receptors (to be discussed further shortly). Another term for these is as globular amyloid β-peptide 1–42 oligomers. This naming emphasizes the primacy of the longer length Aβ-peptide in AD. One of the key findings over the years is the presence of low-n (dimers to octamers) Aβ oligomeric species , especially dimers, in AD tissue samples. As already noted, the model shown in Fig. 8.6 places the dimers in the lead position of the fibrillar (amyloid plaque ) pathway and trimers in the non-amyloidogenic pathway.
8.7 Aβ Serves in a Neurotrophic and Neuroprotective Capacity
Amyloid-β is a synaptic protein, and the earliest and strongest signs of AD take the form of synaptic deficits. Aberrations in synaptic structure and plasticity appear long before deposits of Aβ or tau can be detected and independent of plaque buildup. Aβ is intimately connected to synaptic activity. It is synthesized and released into the extracellular (interstitial) spaces in response to synaptic activity, and localizes to presynaptic and postsynaptic compartments. Once synthesized, Aβ regulates cholinergic and glutamatergic receptor function, and synaptic transmission. These findings support the idea that under normal conditions Aβ has a protective role. It functions as a homeostatic regulator of synaptic plasticity operating through one or more feedback loops to maintain synaptic strength in its proper operating range.
Neural activity stimulates increased production of Aβ through presynaptic and postsynaptic mechanisms. First, depolarization at the presynaptic terminal triggers a Ca2+ influx resulting in an exocytosis of synaptic vesicles. Synaptic vesicle membranes are internalized via clathrin-coated pits result in an increased concentration of APP in lipid raft -bearing endosomes where they come into contact with BACE1 and γ-secretases . Aβ is then released into the extracellular spaces. Secondly, at postsynaptic terminals, NMDA-induced reduction in α-secretase activity occurs furthering the increased production of Aβ. The overall increased production of Aβ leads to synaptic depression and internalization of AMPA receptors resulting in spine loss and rendering the synapses silent. This serves as a negative feedback to throttle back excitatory synaptic transmission that has become too strong.
The amyloid precursor protein and its cleavage products regulate synaptic structure and function at several levels.
In the adult, they
Maintain proper dendritic spine morphology,
Control glutamatergic and cholinergic receptor activity, and
Act as de facto antioxidants through their metal sequestration activities.
During development these proteins and their peptide products
Promote cell–cell and cell–substrate adhesion, and
Facilitate synaptic formation and maturation.
These processes and how they might go awry in AD are now discussed in greater detail.
8.8 Aβ and Synaptic Dysfunction
8.8.1 Synaptic Plasticity at Glutamatergic Synapses
Memories are encoded in the plastic changes in the strengths of the synaptic connections between neurons in the hippocampus and elsewhere in the brain. Memory loss in these regions is the principal sign of AD, and because of that a great amount of attention has been directed at uncovering the molecular machinery behind memory formation and loss at synapses. This machinery has been explored extensively over the past 40 years using laboratory models of synaptic plasticity in test subjects such as mice. These models, most notably long-term potentiation (LTP) in which the synaptic connections are strengthened and long-term depression (LTD) in which they are weakened, are believed to have a general applicability throughout the brain and give basic insights into what is happening in people.
Action potential s trigger neurotransmitter release from the presynaptic axonal terminals and these bind receptors embedded in the postsynaptic density in dendritic terminals and spines, small dendritic protrusions that receive the signals transmitted from axons. A considerable amount of machinery resides in both terminals to supports these actions. On the presynaptic side there is the machinery that ties the arrival of the action potential s to release of the neurotransmitter-loaded vesicles and the machinery that prepares the vesicles and loads them. The postsynaptic side, commonly referred to as the postsynaptic density (PSD) contains a rich assortment of signal receptors, voltage-gated ion channels, signal transducing protein kinases and phosphatases, anchors, adapters and scaffolding proteins, calcium stores, and mRNAs along with the machinery to translate and regulate them.
Three kinds of glutamatergic receptors work together in the hippocampal neurons—N-methyl-d-aspartate (NMDA) receptors, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionate (AMPA) receptors, and metabotropic glutamate receptors (mGluRs). NMDA receptors are rather special in that they are gated both by ligand binding and membrane depolarization. When these ion channels open they permit the entry of Ca2+ ions into the neuron. High levels of NMDA receptor stimulation triggers a signaling cascade resulting in biosynthesis, transport, and insertion of additional AMPA receptors, and to an increase in the number of dendritic spines. The nascent ligand-gated ionotropic (NMDA and AMPA) receptors are transported from the ER to the Golgi and from there travel along the microtubule rail system to dendritic membrane sites. Upon arrival they interact with the actin cytoskeleton and other elements of the PSD. These interactions result in a stronger response to the arrival of neurotransmitter and thus potentiate the signal. Conversely, repeated low levels of NMDA receptor stimulation produce the converse biophysical reaction. The result is depression of the synaptic response to neurotransmitter arrival. Over time, AMPA are internalized and there is a loss of dendritic spines. The former process underlies LTP, while the second kind of modification produces LTD. These processes are modulated by the mGluRs and by nicotinic acetylcholine receptors containing the α7 subunit (α7-nAChR) that are also present and co-localize with the NMDA and AMPA receptors.
Binding of amyloid-β peptides to NMDA receptors influences synaptic transmission in several ways. Expression levels matter and Fig. 8.7 presents an encapsulation of this aspect. At physiologically normal levels Aβ acting at presynaptic and postsynaptic terminals potentiates synaptic transmission. However, if excessive levels of Aβ peptides are present and persist over time they stimulate continued increases in signaling and calcium entry leading to a dangerous excitotoxicity . This sets the stage the internalization and removal of first the NMDA receptors and then the AMPA receptors followed by loss of dendritic spines. These events derail synaptic plasticity and replace it with a persistent depression of synaptic transmission in what is perhaps the earliest stage in the disease. Additional modulatory influences come from the presence of prion PrPC receptors. Binding of Aβ oligomers to these receptors stimulates receptor clustering and activation of additional inappropriate signaling leading to a further excessive entry of calcium, excitotoxicity, and other damaging effects leading to further receptor and spine loss and synaptic failure.
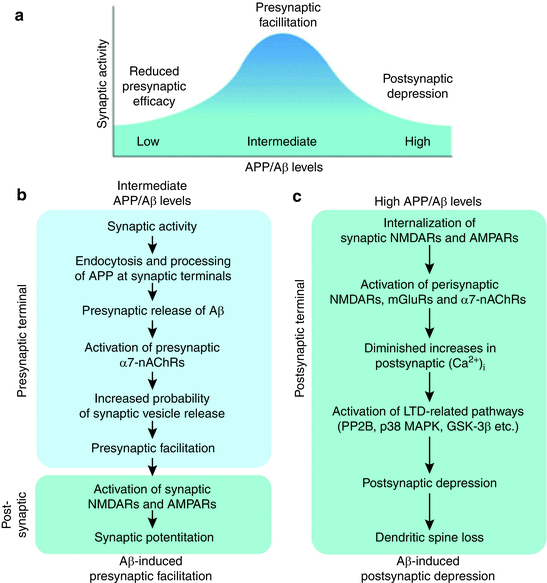
Fig. 8.7
APP acts at pre- and post-synaptic terminals to regulate synaptic transmission Intermediate (normal) levels of Aβ promote proper functioning at synapses whereas levels that are either too high or too low have deleterious effects at the postsynaptic terminal (from Palop Nat. Neurosci. 13: 812–818 © 2010 Reprinted by permission from Macmillan Publishers Ltd)
8.8.2 The Cholinergic System
The study of the cholinergic system dates back to the earliest days of neuroscience. Acetylcholine was, in fact, the first neurotransmitter to be identified. Its discovery and characterization by Henry Dale and Otto Loewi led to their receiving the Nobel Prize in 1936. As noted earlier in this chapter the cholinergic system was an early focus in Alzheimer’s research. Emphasis on this system arose from studies that established that, in Alzheimer’s disease, there was a selective deterioration of cholinergic neurons located in the basal forebrain and that project to the hippocampal region and cortex. This loss was accompanied by a decline in cholinergic neurotransmission and led to development of a number of drugs that treat this deficiency and are still in use today.
Nicotinic acetylcholine receptors containing the α7 subunit are highly expressed in the basal forebrain neurons that project to the hippocampus. These receptors are pentameric ion channels that enable the entry of Ca2+ and Na+ cations. The channels have a high permeability to calcium and as a result influence a variety of calcium-dependent cellular processes. These receptors are present in presynaptic terminals and perisynaptically in the somal/dendritic compartments (see Fig. 8.7). At the presynaptic terminals they modulate calcium homeostasis and the release of acetylcholine, while at the dendritic spines they alter the synaptic currents involved in sensory information processing, learning, memory formation, and protective anti-apoptotic actions. Significantly, these channels function as high-affinity receptors for amyloid-β. In the presence of high levels of Aβ peptides , their normal modulatory and protective roles are lost and instead binding contributes to the destructive hyperexcitability.
8.8.3 Calcium Signaling
Calcium is an ideally suited second messenger, more so than monovalent ions such as sodium or potassium or chloride, and divalent cations such as magnesium. Because of its size and coordination chemistry calcium is well matched to the typical sizes of protein binding cavities, readily forms bonds with proteins, and is able to induce conformational changes. Unlike other second messengers, calcium is not synthesized by cells. Instead, there are two reservoirs of calcium—the extracellular spaces outside the cell and the calcium stores located within the cell. The calcium concentration in the extracellular spaces is on the order of 2 mM, some 20,000 times greater than the resting levels within the cell. Calcium is sequestered within the cell in intracellular stores, regions enriched in calcium buffers located in the lumen of the endoplasmic reticulum, the matrix of mitochondria, and in the Golgi.
The duration of a calcium signal is short. Intracellular calcium levels are restored to their base values fairly rapidly. Buffering agents bind calcium ions before they can diffuse appreciably from their entry point. Free calcium path lengths, the distance traveled by calcium ions before being bound, average less than 0.5 μm, which is far smaller than the linear dimensions, 10–30 μm, of typical eukaryotic cells. In addition to being buffered, ATP-driven calcium pumps located in the plasma membrane rapidly remove calcium ions from the cell, and other ATP-driven pumps transport calcium back into the intracellular stores. The take-up of calcium by buffers, along with its rapid pumping out of the cytosol, produces a sharp localization of the calcium signal both in space and time.
8.9 Amyloid-β and Raft Lipids Regulate One Another
Recall from the last chapter that lipid raft s are cholesterol – and sphingolipid-enriched microdomains . These structures play an important role in aging and Alzheimer’s disease pathology. Lipid rafts are situated in the trans-Golgi and endosomal membranes, and in the plasma membrane of the somal, axonal, and dendritic compartments. In these locations, they provide needed support for the receptors and associated synaptic machinery described in the previous sections. NMDA, AMPA, mGluRs, and α7-AChRs are raft residents as are the amyloid precursor protein and BACE1. In contrast, the α-secretase resides in the plasma membrane in non-raft locations. Targeting sequences possessed by the proteins facilitate their recruitment to the rafts. There, they can carry out their proteolytic, signaling and control functions and regulate the lipid composition of the rafts through positive and negative feedback loops.
The lipid content of the rafts is altered in aged and AD brains. Sphingomyelin (SM) levels are reduced leading to increases in ceramide content. Ceramide is a pro-apoptotic signaling molecule. It is generated through the action of sphingomyelinase (SMase), a hydrolytic enzyme that breaks down sphingomyelin into phosphocholine and ceramide. SMase is activated by cellular stresses and these include not only inflammation but also Aβ peptides , most notably the long Aβ1–42 form. Ceramide stimulates Aβ biogenesis by increasing the half-like of BACE1, thereby setting up a positive feedback loop to drive neurons towards an apoptotic outcome. Under normal operating conditions these actions might protect the brain against defective or excessively stressed neurons, but under aging and AD conditions it establishes ceramide as a potential downstream effector of Aβ toxicity.
The uptake of cholesterol -laden ApoE particles is mediated by LRP1 receptors. The endocytosed lipoproteins are hydrolyzed in lysosomal compartments resulting in the release of their cholesterol content, which then can be incorporated into lipid raft s . The localization of APP to cholesterol-rich rafts is promoted by the presence of a cholesterol-sequestration site on C99, the portion of APP that remains membrane-bound after BACE1 cleavage and release of the ectodomain. Once the second cut is made by the γ-secretase the AICD translocates to the nucleus where it acts at the LRP1/2 promoter to suppress LRP1 expression, thereby throttling back cholesterol uptake. The Aβ peptide, primarily Aβ1–40, acts in concert with these actions by negatively regulating hydroxymethylglutaryl-CoA reductase (HMGR), the main enzyme in cholesterol biosynthesis.
Gangliosides are a prominent family of glycosphingolipid constituents of neuronal lipid raft s . Aβ oligomers bind strongly to gangliosides such as GM1. These interactions sequester Aβ on the rafts, and trigger structural changes. The consequences of these interactions are complex with cholesterol binding an added modulatory factor. Some studies highlight the potential for these interactions to reduce Aβ-oligomer levels in neurons through peripheral clearance, while others emphasize that GM1 may act as a cofactor that seeds Aβ fibrillization. All agree that these interactions along with those involving cholesterol are important.
The cholesterol content of the lipid raft s influences BACE1 and gamma secretase activity. Increases in cholesterol levels promote the association of BACE1 with lipid rafts leading to an increased amyloidogenic processing of APP and a greater production of Aβ. Components of the gamma secretase complex localize to the rafts, as well. These results point to biophysical manipulations of the raft’s lipid components as a way of altering the amyloidogenic processing of APP.
8.10 Amyloid-β Oligomers Initiate Aberrant Signaling
Considerable efforts have been made to identify receptors for Aβo. Finding the correct receptors is made difficult by the sheer variety of oligomeric species and peptide chains that can be produced. One of the putative receptors is PrP. Recall from the previous chapter that the cellular prion protein cannot by itself transduce a signal into neurons but, instead, partners with co-receptors to trigger signaling. The receptors then recruit and activate tyrosine kinases such as Fyn to stimulate further downstream signaling. These pathways can be exploited by Aβo to induce aberrant and potentially toxic signaling cascades at the synapse. An example of how this route can lead to excitotoxicity is depicted in Fig. 8.8a. In this situation, the metabotropic glutamate receptor mGlu5 function as a co-receptor that activates Fyn. That kinase, in turn, phosphorylates nearby NMDA receptor subunits leading to excessive entry of Ca2+ into the postsynaptic terminal.
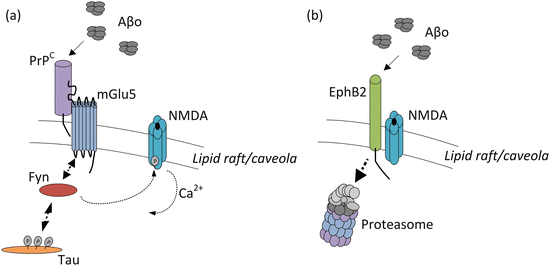
Fig. 8.8
Aberrant signaling by amyloid-β oligomers (Aβo). (a) Signaling initiated by binding to the PrPC/mGlu5/Fyn complex leading to excessive activation of NMDA ion channels and increased excitotoxicity . Mislocalized tau further recruits Fyn to the raft-associated signaling complex resulting in increased phosphorylation and stimulation of NMDA (and other) ion channels. (b) Reduced synaptic plasticity resulting from binding of Aβo to the EphB2 receptors. This action displaces Ephrin ligands; the EphB2 receptors are internalized and degraded by the proteasome, and the NMDA ion channels fail to open when needed
Several candidate receptors for Aβo have been identified. Besides PrPC, ephrinB2 receptors (EphB2) have been found to bind amyloid-β oligomers. The competitive binding of Aβo in place of its normal ligand Ephrin-B2, results in the internalization and degradation of the Eph receptors as shown schematically in Fig. 8.8b. Under normal conditions EphB2 regulates NMDA receptor-mediated calcium entry and by that means regulates synaptic plasticity . The reduction in EphB2 levels impairs that function resulting in inadequate NMDA activity. Another recently identified receptor for Aβ oligomers is the leukocyte immunoglobulin-like receptor B2 (LilrB2). In this situation, Aβo binding initiates a signaling cascade in which the protein phosphatases PP2A and PP2B are stimulated, cofilin is activated, and actin is depolymerized. The result is shrinkage of dendritic spines and loss of synaptic plasticity.
Finally, Aβo can signal through PrPC in more than one way. Another PrPC co-receptor is the low-density lipoprotein receptor-related protein-1 (LRP1). When Aβo binds PrPC/LRP1 it impairs its ability to inhibit the activity of BACE1, one of its normal functions that occur upon entry into the endocytic pathway. This particular activity depends on the presence of a fibrillar/hydrophobic conformation on the part of the Aβ oligomers; soluble/less-hydrophobic oligomeric forms do not bind PrPC as readily. In sum, aberrant Aβo signaling can produce a number of toxic events, most notably, excitotoxicity , receptor malfunction, and dendritic spine loss, all leading to synaptic failure and network breakdown.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


