(Video 29.1). Upper motor neuron signs (hyperreflexia and spasticity) are commonly found in patients with SCA1 and SCA3; cognitive impairment has been reported in association with SCA2, SCA12, SCA13, and SCA17; chorea may manifest in patients with SCA17 or dentatorubral-pallidoluysian atrophy (DRPLA). Saccadic velocity is clearly abnormal in SCA3 and SCA2. Extremely slow saccades are very common in SCA2. Except for tics, all types of movement disorders can been observed in many of the SCAs. Dystonia is mainly associated with SCA17, SCA3, and SCA2. Parkinsonian features can be observed in SCA2, SCA3, or SCA17. Myoclonus has been observed in many of the SCA subtypes, but is most frequent in SCA2 and SCA14.
B. Autosomal-recessive ataxias (ARCAs). ARCAs may present with additional extra–central nervous system signs and symptoms. ARVE deficiency, abetalipoproteinaemia, CTX, and Refsum disease should always be considered in the differential as discussed above. Coenzyme Q10 (CoQ10) deficiency presents with seizures, cognitive decline, pyramidal track signs, and myopathy but may also include prominent cerebellar ataxia. Symptoms may respond to CoQ10 supplementation.
1. Friedreich ataxia (FRDA) is the most common AR ataxia. FRDA is characterized by slowly progressive ataxia with onset usually before 25 years of age associated with depressed tendon reflexes, dysarthria, Babinski sign, and loss of position and vibration sense. About 25% of affected individuals have a presentation with later onset (age >25 years) with retained muscle stretch reflexes and marked cerebellar atrophy on MRI or unusually slow progression of disease.
2. Ataxia telangiectasia (AT) presents with progressive, childhood-onset, cerebellar dysfunction and oculomotor apraxia, frequent infections, choreoathetosis, telangiectasias of the conjunctivae, immunodeficiency, and an increased risk for malignancy, particularly leukemia and lymphoma.
3. Ataxia with oculomotor apraxia type 1 (AOA1) is characterized by childhood onset of slowly progressive cerebellar ataxia followed by oculomotor apraxia that progresses to external ophthalmoplegia associated with severe primary motor peripheral neuropathy and cognitive impairment. Hypoalbuminemia is observed on laboratory investigations.
4. Ataxia with oculomotor apraxia type 2 presents with a similar phenotype as type 1, but age at onset is in the early teens and there is perhaps a lesser degree of certain features. In further contrast to type 1, laboratory studies show normal albumin and high serum α-fetoprotein concentrations.
5. Autosomal-recessive mutations in polymerase g -1 (POLG mutations) are associated with a broad spectrum of CNS and systemic phenotypes, including a mitochondrial recessive ataxic syndrome characterized by cerebellar ataxia, nystagmus, dysarthria ophthalmoplegia, and frequently epilepsy, neuropathy, and dysarthria. Sporadic adult-onset ataxia is most frequently encountered in myoclonic epilepsy associated with ragged-red fibers, mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes (MELAS), and neuropathy, ataxia, and retinitis pigmentosa. Progressive external ophtalmoplegia might point toward the diagnosis of a mitochondrial cytopathy.
C. Associated neurologic signs.
Identifying additional neurologic signs in addition to features of cerebellar dysfunction (dysmetric and saccadic eye movements with nystagmus, dysarthria, a coarse kinetic tremor, dysdiadochokinesia, and a wide-based unstable gait) can provide powerful insight into potential causes of ataxia and dictate secondary investigations).
1. Ataxia and myelopathy.
a. Structural or vascular pontocerebellar abnormalities
b. Inflammatory CNS diseases
c. Multiple sclerosis
d. Superficial CNS siderosis
e. Paraneoplastic syndromes
f. Nutritional deficiencies (vitamin B12, vitamin E, copper)
g. Hereditary conditions including Alexander’s disease, spastic paraplegia 7, autosomal-recessive spastic ataxia of Charlevoix–Saguenay (ARSACS), adult-onset FRDA, or SCA3
2. Ataxia and retinitis pigmentosa/vision loss
a. Refsum disease
b. Vitamin E deficiency
c. SCA7.
d. Mitochondrial disease
3. Ataxia and ocular apraxia
a. AOA1
b. AOA2
c. Whipple’s disease
d. AT
4. Ataxia and chorea
a. Huntington disease
b. SCA17
c. SCA1
d. SCA2
e. DRPLA
f. AT
g. AT type 2
h. Neuroacanthocytosis syndromes
i. Glucose transporter type 1 deficiency
5. Ataxia and tremor
a. FTAX
b. Wilson’s disease
c. SCA12
6. Ataxia and early cognitive impairment
a. CJD
b. GAD ataxia
c. POLG mutations
d. Ataxic variant of SREAT
e. SCA17
7. Ataxia and parkinsonism
a. SCA2
b. SCA3
c. SCA17
d. SCA1
e. MSA
8. Ataxia and polyneuropathy
a. Late-onset Tay–Sachs disease
b. CTX (axonal, demyelinating, or mixed)
c. AT
d. AOA2
e. ARSACS (axonal or demyelinating)
f. Refsum disease (demyelinating)
g. POLG mutations (demyelinating)
h. Late-onset Tay–Sachs disease
i. Several ADSCs including SCA1, SCA2, SCA3, SCA4, SCA8, SCA12, and SCA18.
D. Brain MRI.
Brain MRI is required in the evaluation of all patients presenting with cerebellar ataxia to assess for cerebellar atrophy or structural lesions. MRI is also the method of choice to detect cerebellar malformations, such as Chiari malformation, which can be a cause of sporadic adult-onset ataxia. Global cerebellar atrophy is commonly seen in most causes of chronic inflammatory or degenerative ataxias. Additionally, severe other specific abnormalities can be seen with high-yield diagnostic values as described in previous sections and below (Fig. 29.1).
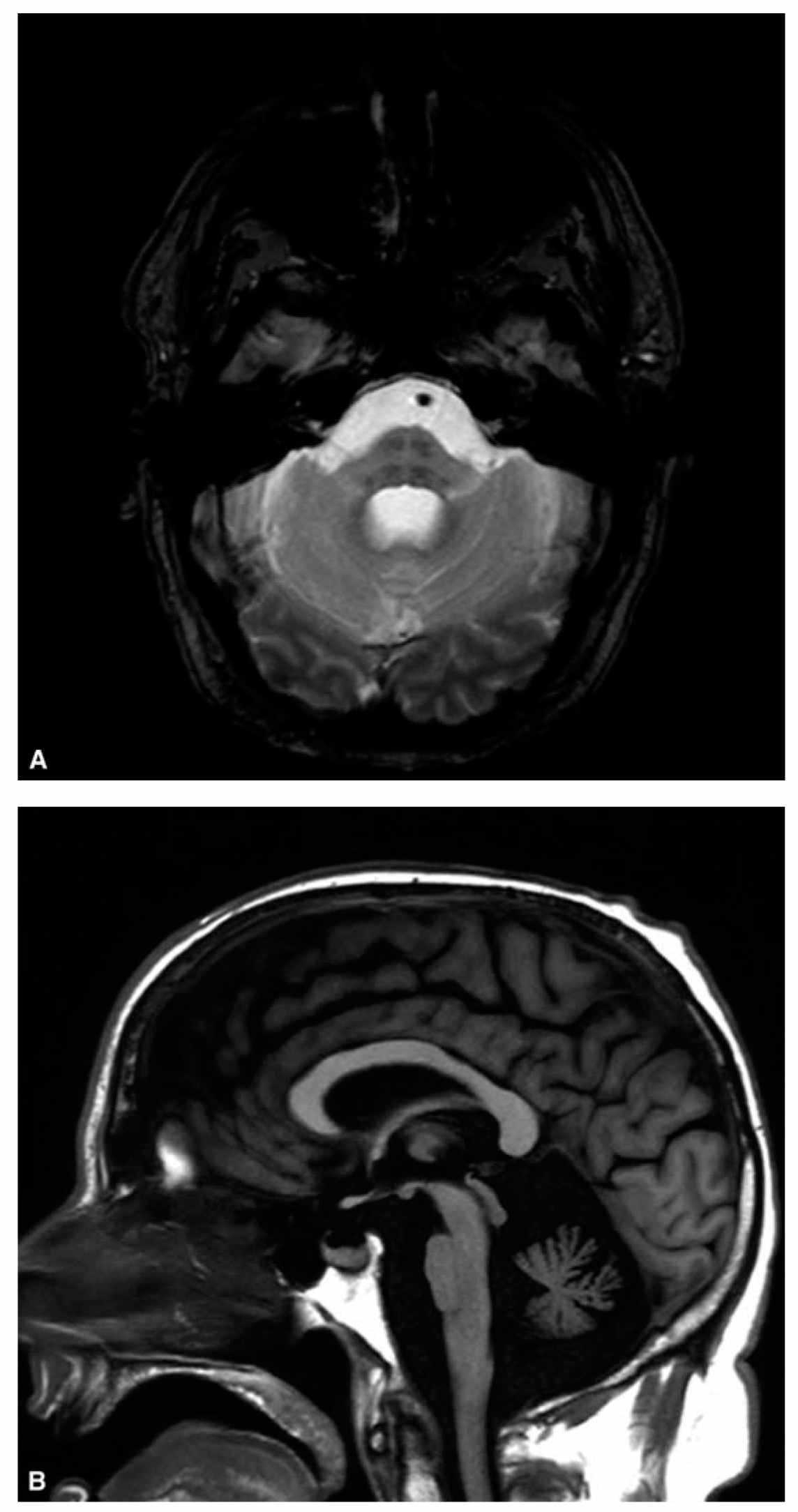
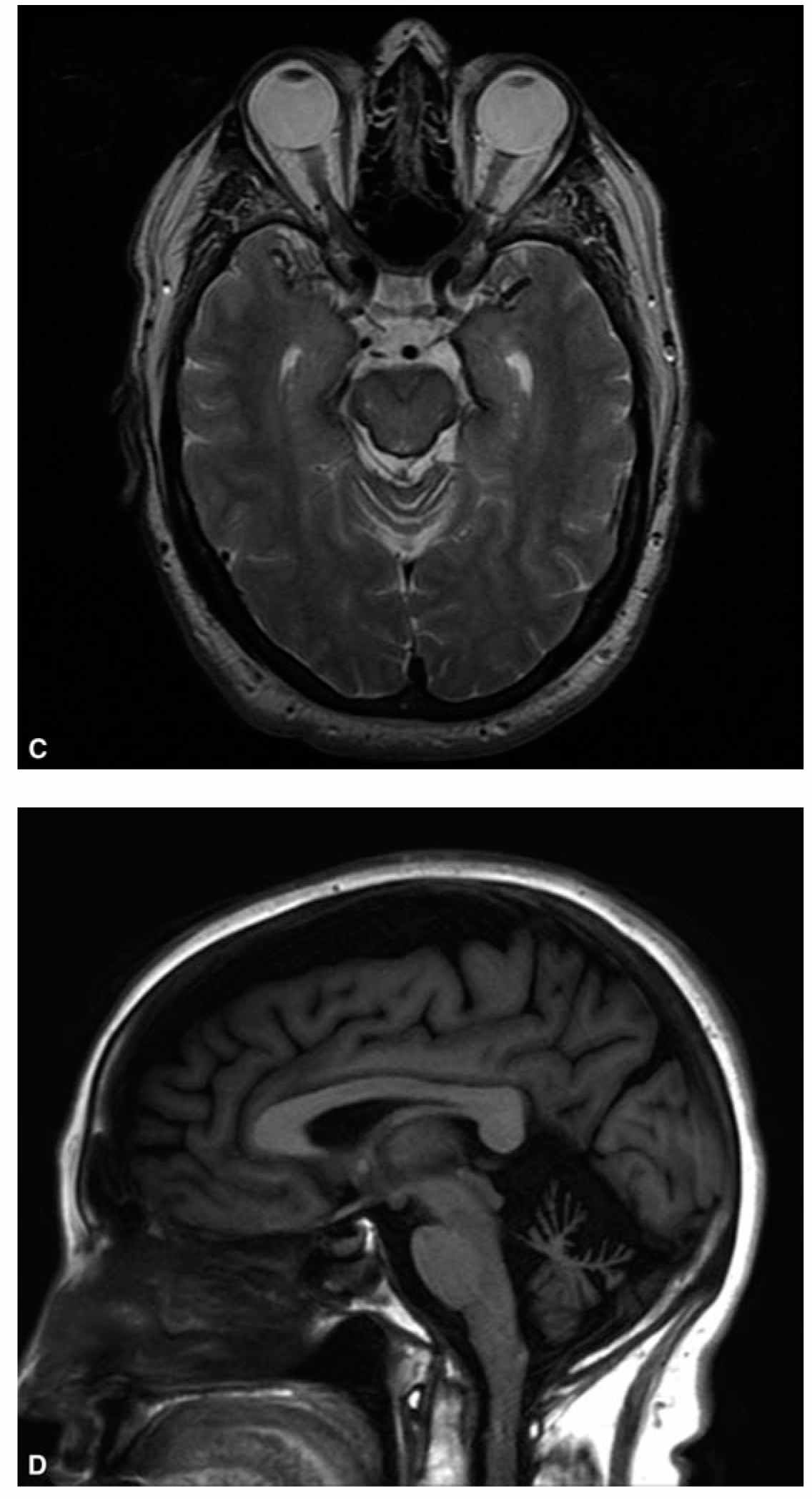
FIGURE 29.1 A shows the “hot-cross bun” sign consisting of a cruciform pattern of hyperintensity in the basis pontis visible on T2-weighted sequences in patients with MSA. B shows marked atrophy of the pons, middle cerebellar peduncles, and cerebellum in the same patient. C shows linear hypointensity on midbrain surface on T2-weighted sequences in a patient with superficial CNS siderosis. D illustrates moderate to severe cerebellar atrophy in a patient with ADCA. ADCA, autosomal-dominant ataxias; CNS, central nervous system; MSA, multiple system atrophy.
1. Cerebellar and cerebral white matter disease hyperintensities
a. Metabolic leukodystrophies
b. Alexander’s disease
c. POLG mutations
e. Adrenoleukodystrophy
f. CTX
2. Severe spinal cord atrophy
a. FRDA
b. Alexander’s disease
3. In patients with suspected AR ataxia, MRI can stratify patients in different categories narrowing the differential and prompting specific testing.
a. FRDA-like phenotype without cerebellar atrophy
(1) FRDA
(2) AVED
(3) Abetalipoproteinemia
(4) Refsum disease
b. FRDA-like phenotype with cerebellar atrophy
(1) Late-onset Tay–Sachs disease
(2) CTX
(3) POLG mutations
(4) Spinocerebellar ataxia with axonal neuropathy
c. Early-onset ataxia with cerebellar atrophy
(1) AT
(2) AOA type 1
(3) AOA type 2
(4) ARSACS
(5) Infantile-onset spinocerebellar ataxia
(6) Marinesco–Sjögren’s syndrome
PRACTICAL APPROACH
A rational strategy for investigating potential etiologies and to dictate further testing is necessary when evaluating patients with cerebellar ataxia. Assessment should start discerning from other causes of unsteadiness including musculoskeletal conditions, vestibular or proprioceptive ataxia, or cognitive dysfunction. At initial visit, the diagnostic evaluation should be directed to in-depth assessment for acquired conditions with hierarchical selection of laboratory testing with focus on potentially treatable conditions. It is important to recognize that in the elderly, multifactorial disease is rather common. Alcohol consumption, exposure to possible toxic agents, chronic infections (including HIV), and history of current or prior malignancies should be obtained. Initial testing should include serum electrolytes and complete blood count, erythrocyte sedimentation rate, antinuclear antibody, Ds-DNA Abs, vitamin B12, methylmalonic acid, homocysteine, thyroid-stimulating hormone, liver enzymes, parathyroid function, vitamin E levels, and serum copper to exclude common metabolic or inflammatory conditions in conjunction with brain MRI. Secondary investigations including lactate and pyruvate, antigliadin antibodies, transglutaminase 2 and 6 antibodies, TPO Abs, paraneoplastic panel, anti-GAD65 Abs, electromyogram/nerve conduction velocities studies, or CSF analysis should be considered based on clinical phenotype.
Additionally, evaluation should focus on obtaining a detailed family history to determine if a hereditary ataxia is present and the suspected mode of inheritance particularly if initial investigations for acquired cases are unrevealing. Age of onset, temporal profile, and associated clinical findings are critical for evaluation. When considering testing for hereditary ataxias, genetic tests should be individualized depending on family history and clinical phenotype. If no acquired cause of the ataxia is identified, the probability is about 13% that the affected individual has SCA1, SCA2, SCA3, SCA6, SCA8, SCA17, or FRDA. If an AR cause of ataxia is possible, testing for elevated α-fetoprotein, serum cholestanol, hypoalbuminemia, or phytanic acid levels should be entertained. Finally, investigating for other rare causes of ataxia including muscle biopsy, CoQ10 in skeletal muscle, amino acids and organic acids in serum or CSF to exclude mitochondrial disease, CoQ10 deficiency, or inborn errors of metabolism can be considered. In cases of late-onset cerebellar ataxia that are found to lack a specific acquired or genetic etiology after complete evaluation, the diagnosis of idiopathic late-onset cerebellar ataxia should be entertained. Clinical exome sequencing is an evolving, newer diagnostic tool that can identify pathogenic gene variants in patients with unexplained ataxia but ancillary testing should be individualized based on clinical presentation.
• The key features of cerebellar dysfunction include dysmetria and saccadic eye movements with nystagmus, dysarthria, action tremors, impaired coordination of targeted and rapid-alternating movements, and a wide-based unstable gait.
• Diagnostic evaluation of ataxia should always include assessment for acquired and causes as acquired and hereditary causes can coexist.
• Age of onset, symptom’s temporal profile, and associated clinical findings are essential elements of ataxia evaluation.
• Brain MRI is required in all patients presenting with cerebellar ataxia to assess for cerebellar atrophy, structural lesions, and high-yield specific abnormalities.
• Initial testing should be individualized based on clinical presentation and aim to exclude common or treatable conditions followed by hereditary and neurodegenerative etiologies.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


