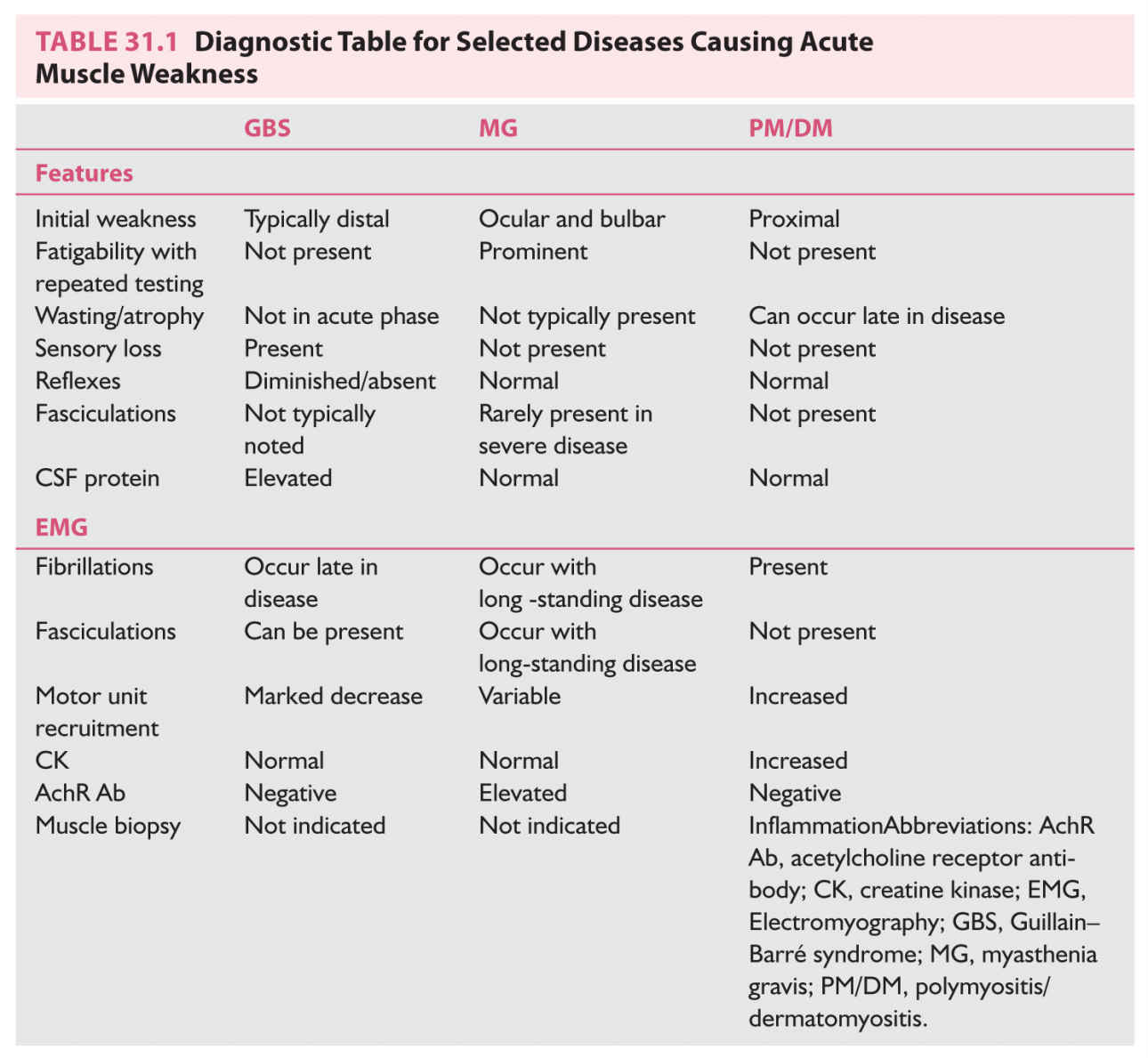
(see Video 31.1). Eventually, dysarthria, dysphagia, and respiratory distress may occur. Ocular myasthenia will present with asymmetric ptosis and diplopia. There is fatigability induced by repetitive exercise. Muscle tone, bulk, reflexes, and sensory examination are normal. Diagnosis is based on clinical examination, single-fiber EMG/repetitive nerve stimulation, and laboratory assessment for acetylcholine receptor antibodies (AchR Abs). AchR-binding antibodies are the most sensitive; blocking and modulating antibodies do not significantly increase the sensitivity. MuSK antibody MG occurs more often in females, with oculobulbar onset.
F. Primary myopathy.
1. PM/DM. Acute inflammatory myopathy usually begins with proximal symmetric weakness involving the muscles of the shoulder and hip girdle. Muscle tone, bulk, and muscle stretch reflexes are normal. There are no sensory deficits. PM is usually painless or may have an achy nonspecific muscular pain. Consider DM if typical skin lesions are present (erythematous rash in the periorbital, malar, forehead, or chest region, and a scaly, erythematous rash over the knuckles and extensor surfaces). Serum creatine kinase (CK), lactic acid dehydrogenase (LDH), and aspartate aminotransferase (AST) levels are often elevated. Erythrocyte sedimentation rate (ESR) may be increased. NCS and amplitude are normal. Needle EMG will show a myopathic pattern in affected muscles. Muscle biopsy shows an inflammatory response, which differs depending on the pathologic process present: perimysial inflammation is noted in DM and intrafascicular inflammation is present in PM. The biopsy will also show muscle fiber necrosis and a variable degree of muscle fiber regeneration.
2. Rhabdomyolysis occurs after severe injury to muscles: it may be focal or generalized depending upon the injury. Patients will have swelling and pain in the affected muscles, demonstrate weakness, and have markedly elevated CKs. Acutely elevated CKs may put the patient at risk for renal failure and focal injury may cause compartment syndrome. These patients need to be hospitalized for monitoring and careful hydration.
3. Acute infectious myositis. Postviral myositis is associated with myalgia and weakness, which may be severe. HIV infection can manifest as proximal muscle weakness.
4. Acute toxic myopathy. Most drug-induced myopathies are subacute in onset. Amiodarone can cause acute myopathy and paralysis. Hyperthyroidism can cause acute weakness in severe cases.
5. Acute periodic paralysis is a group of primary muscle diseases associated with acute transient quadriparesis without respiratory compromise. Patients may have had attacks for years but will present to the emergency room for a particularly severe attack. The episodes of quadriparesis typically resolve spontaneously, although, over the years, patients may develop a chronic myopathy/weakness. These diseases are also known as channelopathies because the etiology is a defect in an ion pore of the muscle membrane. Hyperkalemic periodic paralysis (Hyper PP) is caused by a defect in the gene coding for a muscle sodium channel (SCN4A) and hypokalemic periodic paralysis (Hypo PP) is caused by either a gene defect in a calcium channel of the muscle (CACNA1S) or the sodium SCN4A channel. There is significant overlap between the presentations of both conditions and diagnosis is best confirmed by genetic testing. Patients with Hyper PP may have clinical myotonia. The diagnosis is suspected if the patient has a history of intermittent weakness induced by exertion or a high-carbohydrate diet, a family history, and abnormal serum potassium levels during attacks. EMG/NCS may be normal or may show decreased compound muscle action potentials (CMAPs); the prolonged exercise test can be used to demonstrate a reduction in CMAP amplitude. Muscle biopsy may show a vacuolar myopathy.
6. Acute critical illness myopathy occurs in the ICU setting. Patients will have flaccid paralysis and difficulty weaning from the ventilator. Sometimes, this disease is associated with treatment for status asthmaticus using high-dose steroids and neuromuscular blockade agents. The EMG shows either myopathic features or an electrically silent muscle; it is differentiated from ICU neuropathy by normal sensory NCS. Muscle biopsy typically shows loss of myosin filaments at electron microscopic examination. Patients may recover from this process, unlike acute critical illness neuropathy.
LABORATORY STUDIES
A. Blood tests. If myositis is suspected, measurement of serum CK, ESR, LDH, and AST is useful. Anti-Jo antibody test results are positive in approximately 30% of cases of PM. The presence of this antibody is a marker of risk for pulmonary fibrosis. Other autoantibodies associated with inflammatory myopathy are insensitive and not diagnostically useful.
If vasculitis is suspected, measure ESR, serum complement, antinuclear antibodies, antineutrophil cytoplasmic antibodies, and cryoglobulins. Consider evaluating the patient for HIV and chronic hepatitis infection.
If MG is suspected, check AchR Ab (binding) titers and thyroid function tests. If these results are negative, consider testing MuSK antibodies and antibodies to the voltage-gated calcium channel (Table 31.1).
In conditions such as periodic paralysis, serum potassium and thyroid function tests may or may not be helpful. Potassium (K+) levels can fluctuate greatly during the course of an attack. The genetic defects for some channelopathies are known. Commercial testing is available for the genes encoding for the SCN4A channel (Hyper PP/paramyotonia congenital and Hypo PP, type 2) and the CACNA1S channel (Hypo PP, type 1).
Patients affected with AMAN may have had a recent Campylobacter jejuni infection. The anti-GQ1b ganglioside antibody is both sensitive and specific for the Miller Fisher variant of GBS. This variant manifests as ophthalmoparesis, ataxia, and areflexia.
In suspected cases of West Nile viral infection, acute and convalescent titers should be drawn (serum IgG and IgM levels).
B. Lumbar puncture is indicated in the evaluation of patients with suspected GBS, in which CSF may show protein elevation with minimal or absent pleocytosis (albuminocytologic dissociation). In patients with an acute poliomyelitis picture (acute MND), the lumbar puncture may show lymphocytic pleocytosis and protein elevation. Check for WNV-IgG antibodies, and, if the patient is immunocompromised, check the CSF for herpes simplex virus, varicella zoster virus, and CMV infections as well.
C. Electrodiagnostic studies. EMG and NCS are extremely useful in the evaluation of disorders of motor neurons, peripheral nerves, neuromuscular junctions, and muscles. The value of electrodiagnostic tests is discussed in Chapter 33.
D. Muscle biopsy. Muscle tissue can be obtained by means of open incision. The site of muscle biopsy should involve a weak but not atrophic muscle. Specimen handling and interpretation of muscle biopsy findings by an experienced pathologist are crucial. Muscle biopsy aids in diagnosis of acute primary muscle pathologies such as PM/DM.
E. Nerve biopsy is most often performed on the sural nerve. This procedure should be performed only when the biopsy results will influence management. One of the leading indications for nerve biopsy is the suspicion of vasculitis (mononeuritis multiplex).
F. MRI of the affected muscles may indicate inflammation in an infectious/inflammatory myositis. Atrophy of the muscles will be noted in subacute to chronic processes, such as dystrophy or MND.
DIAGNOSTIC APPROACH
Diagnosis begins with establishing the presence of weakness and then determining whether the weakness reflects upper or lower motor neuron involvement. After exclusion of upper motor neuron weakness, further localization within the motor unit is needed. Diagnosis often requires support by laboratory studies. Elevated CK is the easiest way to identify muscle inflammation. An EMG/NCS will further localize the lesion. Muscle biopsy is recommended for evaluation of PM/DM. Nerve biopsy is indicated mainly in cases of suspected vasculitic neuropathy.
MANAGEMENT
Patients with acute onset of generalized neuromuscular weakness need to be hospitalized, particularly those with acute paralysis or paresis. If respiratory or bulbar muscles are compromised, patients need admission to an ICU. Bedside pulmonary function tests (FVC and NIF) are used to monitor respiratory function. A sustained drop in FVC, or an FVC <1 L, indicates impending respiratory failure, and intubation is indicated. Neuromuscular diseases with a subacute onset may sometimes be managed in the outpatient setting. Many of the therapies for neuromuscular weakness require careful monitoring and follow-up assessment, especially if steroids and immunosuppressive agents are used.
Key Points
• Muscular weakness must be distinguished from fatigue, lethargy, and tiredness, which are not neuromuscular in origin.
• Physical examination should focus on determining the pattern of weakness; localizing the deficit along the motor unit; and assessing for sensory involvement.
• CK testing will assess for muscle inflammation.
• Patients with respiratory or bulbar involvement and those with acute rhabdomyolysis need to be hospitalized and monitored carefully.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


