Atypical Absence, Myoclonic, Tonic, and Atonic Seizures
William O. Tatum IV
Kevin Farrell
Atypical absence, myoclonic, tonic, and atonic seizures are all types of generalized seizures that occur when the initial clinical and ictal electroencephalographic changes arise from both hemispheres (1). Atypical absence, tonic, atonic and often myoclonic seizures are symptoms of an underlying disease process (1, 2, 3, 4). Despite an apparent homogeneous classification, various pathophysiologies may occur. Conversely, although several seizure types can coexist, they may share a common epileptogenic substrate (5). Classification of symptomatic seizures has been recognized since 175 AD (6). Atypical absence, myoclonic, tonic, and atonic seizures are often associated with mental retardation, an abnormal electroencephalogram (EEG), a poor response to therapy, and typify patients with encephalopathic generalized epilepsy (7). Some seizures that appear generalized clinically may be focal on the ictal EEG (8), emphasizing the importance of both the clinical features of the seizure and the EEG in classification (8,9). Seizures may defy classification, because this population often has comorbid mental retardation and multiple handicaps that limit subjective reporting and objective behavioral description. Videoelectroencephalographic monitoring has improved recognition, identification, and classification for patients with atypical absence, myoclonic, tonic, and atonic seizures (10), with revision of former classifications (11, 12, 13) and the development of newer semiologic-based classifications (14).
SEIZURE TYPES
Atypical Absence Seizures
Clinical Features
Absence seizures are subdivided into typical and atypical forms. The initial description of atypical absence seizures was in association with atypical, or slow spike-and-wave (SSW), EEG discharges that were less than 3 Hz (15,16). Petit mal is a colloquialism used to describe typical absence seizures (17), which may be either simple or complex. The semiology of atypical absences may resemble that of complex typical absence, although atypical absences are usually differentiated by the association with a severe epileptic encephalopathy, with mental retardation, multiple seizure types, neurologic disabilities, and EEG features (16,18). Atypical absence seizures may occur at any age, but they rarely begin before 2 years of age or after the teenage years (19). Such seizures may occur alone with brief staring and a variable degree of impaired consciousness, or may be prolonged with atypical absence status. Behaviorally, they are often associated with motor signs, particularly changes in muscle tone, but also including clonic components and autonomic features (16,19). Atypical absence seizures evolve gradually, with less abrupt onset and termination, more pronounced changes in tone, and durations that, unlike typical absence seizures, last longer than 5 to 10 seconds and possibly for minutes. Consciousness is variably impaired, and postictal confusion is brief when observed (2,16). Atypical absence seizures are most likely to occur in states of drowsiness, and do not show activation with hyperventilation and photic stimulation (18). Nonepileptic staring episodes may mimic atypical absence seizures in patients with Lennox-Gastaut syndrome (LGS) (20). In one study, episodes of staring were found to be epileptic in origin in only 27% of patients on video-electroencephalographic monitoring (21), with a more reliable diagnosis of seizures established when motor features were observed.
Electrophysiology
Atypical absence seizures have a characteristic pattern on electroencephalography (22), with interictal generalized SSW discharges that are often irregular, asymmetrical, and lower in amplitude, with spikes (or sharp waves) that repeat at approximately 1.5 to 2.5 Hz (Fig. 20.1). This is in contrast to the regular generalized 3-Hz SSW pattern associated with typical absence seizures. The EEG background is usually diffusely slow. Focal or multifocal independent epileptiform discharges or even generalized polyspike-and-wave activity may appear concomitantly. The individual SSW discharges may occur as single complexes or in bursts that are longer in duration and more blunted in morphology in encephalopathic generalized epilepsy (15,19). The SSW pattern on EEG is typically a malignant pattern, although the appearance is not always associated with a change in behavior. Patients with SSW discharges on EEG and a cryptogenic etiology are more likely to show bilaterally symmetrical SSW activity than those with lateralized structural abnormalities, who show asymmetrical EEGs with higher-amplitude SSW discharges over the unaffected hemisphere (23).
The ictal EEG associated with atypical absence seizures typically demonstrates diffuse, irregular SSW discharges with or without lateralization; however, irregular diffuse fast activity at 10 to 13 Hz, or a combination of fast spike wave or sharp waves of increasing amplitude, may also be seen (16). Forms of absence seizures “intermediate” between typical and atypical absences have been described, with cognitive impairment, social and learning handicaps, pharmacoresistance, poorer prognosis, and fast rhythmic discharges during sleep (similar to those seen in patients with LGS). Sleep may help identify rhythmic fast activity on EEG that is the marker for this transitional form (24). Centromedian thalamic nuclei recording through implanted intracerebral electrodes during atypical absence seizures has shown simultaneous 1- to 2-Hz SSW complex discharges (25). Quantitative electroencephalography has elucidated interhemispheric asynchrony and morphologic asymmetries of SSW discharges to help differentiate generalized epilepsy from frontal lobe epilepsy with secondary bilateral synchrony (26). A selective effect on electroencephalography using antiepileptic drugs (AEDs) to modify the atypical SSW pattern underlying atypical absence seizures has been reported (27).
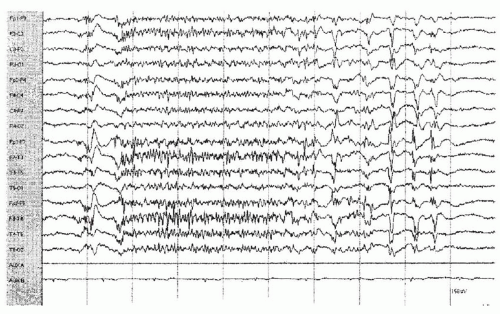 Figure 20.1 Generalized paroxysmal fast activity preceded by polyspike-and-wave discharges and followed by slow spike-wave activity in a 28-year-old male with secondary generalized epilepsy, mild static encephalopathy, and tonic seizures. The burst of 14 to 18 Hz, followed by slow spike-wave activity, was asymptomatic. |
Clinical Correlation
A spectrum of clinical and electroencephalographic manifestations of absence seizures has been noted (28) and further delineated by video-electroencephalographic monitoring (29). Although atypical absences are one of the most common seizure types observed in patients with LGS, they have received considerably less attention than typical absence seizures (30). In studies of longer than 10 years’ duration, patients with LGS and a predominance of atypical absence seizures have demonstrated a strong clinical correlation with poor seizure outcome (31). Patients with combined seizure types, including those with epilepsy who have myoclonic absences with a prominent myoclonic component to the absences, carry a guarded prognosis despite the associated 3-Hz SSW pattern that typically denotes a favorable prognosis. Distinguishing atypical absence from complex typical absence seizures may be challenging, although the clinical course and seizure types seen with encephalopathic generalized epilepsy, with a paucity of automatisms, tonal changes, and longer seizure duration, may help. Complex partial seizures may manifest as brief staring attacks and mimic atypical absences,
but the clinical course with focal clinical and EEG features should help distinguish the two (32). Secondary bilateral synchronous SSW discharges on EEG may occur with focal epilepsy of frontal lobe origin, yet even in the presence of a focal pathologic process, atypical absence seizures are characterized by their association with generalized clinical and electrographic abnormalities (33, 34, 35). Atypical absences may overlap with SSW complexes during slow sleep (36), and occur with the Landau-Kleffner syndrome, the syndrome of continuous spike waves during slow sleep, atypical benign partial epilepsy, and electroencephalographic status of benign Rolandic epilepsy (37).
but the clinical course with focal clinical and EEG features should help distinguish the two (32). Secondary bilateral synchronous SSW discharges on EEG may occur with focal epilepsy of frontal lobe origin, yet even in the presence of a focal pathologic process, atypical absence seizures are characterized by their association with generalized clinical and electrographic abnormalities (33, 34, 35). Atypical absences may overlap with SSW complexes during slow sleep (36), and occur with the Landau-Kleffner syndrome, the syndrome of continuous spike waves during slow sleep, atypical benign partial epilepsy, and electroencephalographic status of benign Rolandic epilepsy (37).
Myoclonic Seizures
Clinical Features
A sudden, brief, involuntary, shock-like muscular contraction that results in a body movement is referred to as myoclonus (38,39) and may be epileptic (myoclonic jerks) or nonepileptic. Myoclonic seizures are generalized seizures (9,11) that occur either as part of an idiopathic epilepsy syndrome or in encephalopathic generalized epilepsy. When combined manifestations occur, such as myoclonic absence, semiology is identified by the primary component with absence the main seizure type. Four types of epilepsy with myoclonic components have been described (40)—combined myoclonic seizure types (Chapter 31), myoclonic seizures associated with encephalopathic generalized epilepsy syndromes (Chapter 26), myoclonic seizures associated with idiopathic generalized epilepsy syndromes (Chapter 25), and infantile spasms (Chapter 21)—and are discussed elsewhere in this text. Infantile spasms may have a shock-like appearance, although they have dissimilar EEG correlates than myoclonic seizures. Massive epileptic myoclonus implies that a bilateral jerk is large enough to create a fall. Whereas myoclonic seizures usually imply a generalized onset, cortical reflex myoclonus is a term that reflects a motor movement as a result of focal epilepsy (41), may appear segmental, and may originate in the regions of the brain responsible for motor activation. Reticular reflex myoclonus, on the other hand, occurs with generalized epilepsy but originates in subcortical structures of the brainstem.
Myoclonic seizures are characterized by sudden, involuntary, brief, muscle contractions of the head, trunk, or limbs. They usually occur without detectable loss of consciousness and may be generalized, regional (involving two adjacent areas), or focal (confined to one area). They may be regular or erratic, symmetrical or asymmetrical, synchronous or asynchronous, and positive or negative (41). Myoclonic seizures are often bilateral jerks that vary from subtle restricted twitches of the periocular or facial muscles to massive movements, with generalized involvement of the arms and legs accompanied by falling or retropulsion (2). They may occur singly or in repeated clusters, with some cognitive impairment noted during prolonged clusters or myoclonic status epilepticus (2). The associated features, rather than the semiology of the myoclonic seizures themselves, define the syndromes associated with myoclonic seizures.
Electrophysiology
In general, myoclonic jerks associated with encephalopathic generalized epilepsy have a high-amplitude, bisynchronous SSW or polyspike-and-wave discharge as the electrophysiologic correlate (Fig. 20.2). A brief latency between short bursts of synchronized electromyographic potentials in agonist and antagonist muscles, and that of the corresponding spikes, occurs. The spikes are time-locked events that are coupled with the myoclonic jerks that follow. By using back-averaging techniques, latencies are found to occur between 21 and 80 milliseconds (42,43). When a myoclonic jerk is generated by subcortical structures, a generalized spike discharge follows the first electromyographic sign of myoclonus; however, in this case, an epileptogenic phenomenon is disputed by some (43). Negative myoclonus caused by a lapse of tone, which can be seen only during antigravity posture, is coupled with either the slow wave or the second positive component of a polyspike-and-wave discharge (43). Myoclonic seizures have correlates with an electromyographic pattern, demonstrating a brief synchronous potential of less than 50 milliseconds that is seen simultaneously in the involved muscle groups (41). During the jerks, medium- to high-amplitude repetitive 16-Hz spikes are seen. (44,45). The background activity of the interictal EEG in patients with encephalopathic generalized epilepsy is characteristically diffusely slow. A unique EEG pattern is seen in early myoclonic encephalopathy and neonatal myoclonic seizures, with burst suppression or multiple paroxysmal abnormalities with random asynchronous attenuations (46). Recordings from thalamic nuclei during myoclonic seizures demonstrate subcortical slow polyspike-and-wave discharges that lead those recorded on the scalp surface in patients with LGS (25,47). Giant visual evoked potentials appearing as occipital high-amplitude polyphasic spikes may be observed during intermittent photic stimulation at less than 3 Hz in patients with neuronal ceroid lipofuscinosis of late infancy, in which myoclonic, as well as atypical absence and atonic, seizures are common (48).
Focal myoclonus is suspected to be derived from a hyperexcitable cortex responsible for corresponding motor activation. A recordable paroxysmal depolarizing shift in animal models may occur with cortical application of proconvulsants (49). Electrographic secondary bilateral synchrony has been reported in patients with myoclonic and partial seizures (50), with generalized epileptiform discharges showing a small delay in interhemispheric propagation as a function of coherence and phase analysis, suggesting frontal lobe onset (50).
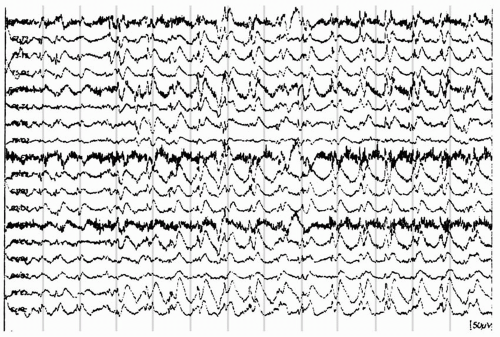 Figure 20.2 A burst of slow spike-and-wave discharges in a 9-year-old girl with secondary generalized epilepsy and atypical absence seizures. Note the 1.5-Hz frequency and intermittent left lateralization that is seen. |
Clinical Correlation
Most epilepsies with myoclonic seizures begin during the first 5 years of life (51). They are clinically and etiologically heterogeneous, and represent a group of disorders that may occur in many epilepsy types and syndromes from early infancy into adulthood (52,53). Myoclonic seizures must be differentiated from infantile spasms, partial seizures with tonic posturing, and nonepileptic conditions (54), and difficulty may arise in differentiating massive myoclonic seizures from tonic and atonic seizures. Causes of myoclonic seizures vary greatly from acquired causes of almost any etiology to familial epilepsies with varied inheritance patterns (52,53). There are subgroups of patients with idiopathic generalized epilepsy with refractory myoclonic seizures and developmental delay, yet with a genetic component. Myoclonic-astatic epilepsy and severe myoclonic epilepsy in infancy represent the more severe phenotypes of generalized epilepsy with febrile seizures plus (GEFS+), which mimic patients with encephalopathic generalized epilepsy, but with no discernible etiology and a genetic foundation for expression (55). Epilepsy with myoclonicastatic seizures is a syndrome intermediate between idiopathic and encephalopathic generalized epilepsy, with febrile seizures and subsequent myoclonic jerks during childhood that involve mainly the axial muscles, more than the face, upper trunk, and arms with jerks strong enough to cause patients to fall (i.e., astatic seizures) (56,57). Similarly, epilepsy with myoclonic absences is characterized by prolonged absence seizures with prominent rhythmic generalized myoclonic jerks involving both shoulders, arms, and legs, which may repeat at 3 Hz during activation techniques, distinguishing myoclonic absence from childhood absence epilepsy (36,58).
A symptomatic or cryptogenic etiology is often found when myoclonic seizures begin before the age of 4 years (55). Early myoclonic encephalopathy manifests as neonatal-onset irregular myoclonic jerks. Severe myoclonic epilepsy of infancy (Dravet syndrome) occurs with myoclonic seizures following febrile seizures during the first year of life (59). Myoclonic seizures are common but least characteristic in patients with LGS, although a myoclonic variant that has a better prognosis for cognitive development has been noted (60). The progressive myoclonus epilepsies (see Chapter 26) are a rare but extremely debilitating and progressive heterogeneous subgroup of encephalopathic generalized epilepsy, with myoclonic
seizures as the clinical marker (61). The presence of mental retardation and abnormal neurologic examination does not preclude an independent idiopathic generalized epilepsy (62), but severe myoclonic epilepsy of infancy, early myoclonic encephalopathy, LGS, the progressive myoclonus epilepsies, and mitochondrial encephalopathies are strongly associated with myoclonic seizures and portend an unfavorable prognosis. Myoclonic status epilepticus typically occurs in patients with encephalopathic generalized epilepsy (63,64), although it occurs less frequently during sleep (65).
seizures as the clinical marker (61). The presence of mental retardation and abnormal neurologic examination does not preclude an independent idiopathic generalized epilepsy (62), but severe myoclonic epilepsy of infancy, early myoclonic encephalopathy, LGS, the progressive myoclonus epilepsies, and mitochondrial encephalopathies are strongly associated with myoclonic seizures and portend an unfavorable prognosis. Myoclonic status epilepticus typically occurs in patients with encephalopathic generalized epilepsy (63,64), although it occurs less frequently during sleep (65).
Tonic Seizures
Clinical Features
Tonic seizures are generalized seizures (12,13,66) that are convulsive (13,67), but may be very brief, appear non-convulsive, and are grouped with absence, myoclonic, and atonic seizures (2). The prevalence is inadequately represented, given the discrepancy between observed seizure incidence and seizures noted on electrographic recording during video-electroencephalographic monitoring (10). In an effort to distinguish different forms of tonic seizures, the taxonomy includes (a) tonic axial seizures with abrupt tonic muscular contraction and rigidity of the neck, facial, or masticatory muscles; (b) global tonic seizures involving widespread contraction of the axial and appendicular musculature; and (c) tonic axorhizomelic seizures as an intermediate form, with contraction of the upper limb muscles and deltoid muscles that leads to elevation of the shoulders. Partial seizures with asymmetrical tonic posturing are referred to as tonic postural seizures. Short tonic seizures have high-amplitude, rapid muscular contractions maximal in the neck and shoulder girdle, but involve mainly the axis of the body and trunk and last 500 to 800 milliseconds, with forward leaning that resembles infantile spasms (68). When tonic posturing is observed in patients with West syndrome and infantile spasms, tonic spasms with massive flexion of the head, trunk, and extremities (“Jackknifing”) is a term used to describe a seizure type that is refractory to treatment (69,70). Myoclonic jerks may appear to be brief tonic seizures but are actually single muscular contractions lasting less than 200 milliseconds. Tonic seizures, on the other hand, are more sustained, lasting seconds (71) with increasing intensity, although they may be associated with an atonic or myoclonic component (25). Prolonged tonic seizures with a vibratory component may resemble generalized tonic-clonic seizures (72), although tonic seizures are briefer, averaging 10 to 15 seconds (71).
 Figure 20.3 A 32-year-old female with cryptogenic Lennox-Gastaut syndrome and brief, massive tonic seizures. Note the large scar on her forehead from a propulsive fall associated with a tonic seizure. Also, note the left chest surgical scar at the site of vagus nerve stimulator placement. |
Tonic seizures may vary, from a short, upward deviation of the eyeballs with or without oscillatory nystagmus to a more intense seizure, with generalized symmetrical or asymmetrical tonic stiffening, loss of consciousness, and falls and repeated injury (10,16). Falls may be forward or backward, depending on whether the axial and lower limb musculature are fixed in flexion, or less commonly with extension of the head and/or trunk (16). Scars from old injuries may be observed on the forehead (Fig. 20.3) and occipital regions, reflecting injury patterns associated with seizures. Contraction of the respiratory and abdominal muscles may create a high-pitched cry or period of hypopnea. Seizure intensities may vary among patients and individuals, and combined seizure types may occur (73). The duration of tonic seizures is several seconds to a minute, although most last for 5 to 20 seconds and are most common during stages I and II of nonrapid eye movement (NREM) sleep (10). Autonomic signs include respiratory, heart rate, or blood pressure increases; pupillary dilation; and facial flushing (10). Postictally, patients have variable degrees of cognitive and motor recovery (74), with the depth of the postictal
state proportional to the seizure intensity (10). In patients with LGS, tonic seizures are reported to occur in large numbers during NREM sleep (75). Tonic status epilepticus (75) may occur in 54% to 97% of patients with an insidious or brief initial tonic component (71).
state proportional to the seizure intensity (10). In patients with LGS, tonic seizures are reported to occur in large numbers during NREM sleep (75). Tonic status epilepticus (75) may occur in 54% to 97% of patients with an insidious or brief initial tonic component (71).
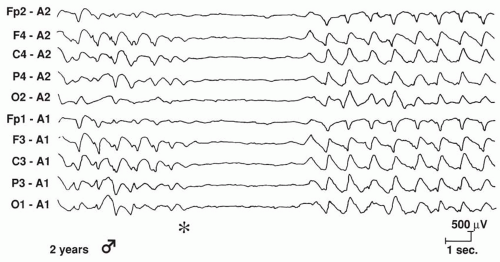 Figure 20.4 An electroencephalogram that demonstrates an abrupt, generalized “flattening” during a tonic seizure in a 2-year-old boy. |
Electrophysiology
Nonepileptic tonic posturing is readily distinguished by normal interictal and ictal EEGs (76,77). The interictal EEG characteristics seen with tonic seizures are dependent on the specific epileptic syndrome. In most patients, tonic seizures are associated with encephalopathic generalized epilepsy, and a diffusely slow background with multifocal spikes and sharp waves on EEG is suggested (78). In patients with LGS beginning before the age of 5 years, generalized SSW complexes may not appear until the onset of epilepsy is well established (79). The ictal EEG manifestations of tonic seizures associated with encephalopathic generalized epilepsy reveal a generalized frontally predominant initial attenuation of background activity that is associated with desynchronization (Fig. 20.4) and precedes the bilateral 10- to 25-Hz spikes, with the amplitude ranging from flattened scalp EEG to greater than 100 μV (80). Generalized paroxysmal fast activity (Fig. 20.5), which
consists of diffuse, repetitive, medium- to high-amplitude spike discharges, occurs as the counterpart of tonic seizures during slow-wave sleep (81). Mixtures of clinical and electroencephalographic patterns may be seen, with tonic-absence seizures occurring with a tonic seizure and subsequent staring associated with generalized paroxysmal fast activity, followed by SSW, on EEG (Fig. 20.6) (73). Fast ictal spike discharges were noted in deeper centromedian thalamic nuclei, correlating bilaterally with the onset of tonic seizures and diffuse scalp EEG changes in patients with LGS (25). Rhythmic ictal theta and delta patterns that differ from the background activity have been described in patients with tonic status epilepticus (82), and non-convulsive status is not an uncommon occurrence in patients with LGS (83).
consists of diffuse, repetitive, medium- to high-amplitude spike discharges, occurs as the counterpart of tonic seizures during slow-wave sleep (81). Mixtures of clinical and electroencephalographic patterns may be seen, with tonic-absence seizures occurring with a tonic seizure and subsequent staring associated with generalized paroxysmal fast activity, followed by SSW, on EEG (Fig. 20.6) (73). Fast ictal spike discharges were noted in deeper centromedian thalamic nuclei, correlating bilaterally with the onset of tonic seizures and diffuse scalp EEG changes in patients with LGS (25). Rhythmic ictal theta and delta patterns that differ from the background activity have been described in patients with tonic status epilepticus (82), and non-convulsive status is not an uncommon occurrence in patients with LGS (83).
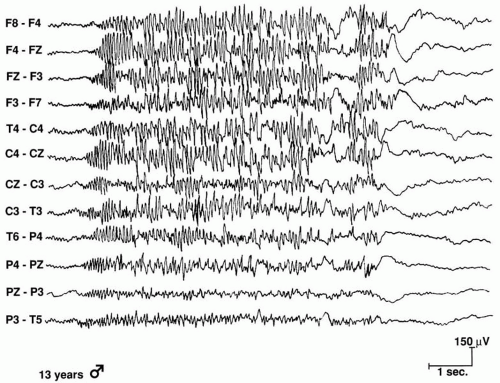 Figure 20.5 Generalized paroxysmal fast activity with bilateral 15 Hz of increasing amplitude during a tonic seizure in a 13-year-old girl. |
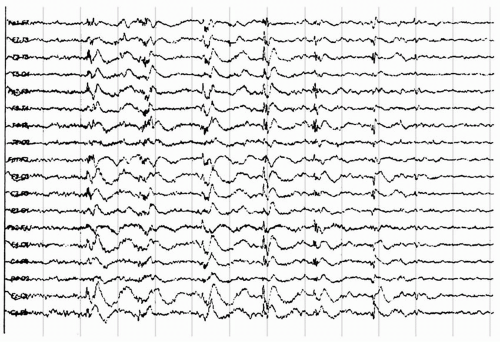 Figure 20.6 The electroencephalogram of a 27-year-old patient with secondary generalized epilepsy, static encephalopathy, and myoclonic seizures demonstrates generalized polyspike discharges with intermittent mild myoclonus of the neck and proximal upper limbs.
Related posts:Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|



