Autoimmune and Postinfectious Diseases
Agustín Legido
Silvia N. Tenembaum
Christos D. Katsetos
John H. Menkes
This chapter considers several groups of neurologic diseases believed to result from a failure of the normal mechanisms of self-tolerance. One group consists of the primary demyelinating diseases of the central nervous system (CNS), the second the immunologically mediated diseases affecting CNS gray matter, the third the immunologically mediated demyelinating diseases of the peripheral nervous system, and the last the primary and secondary systemic vasculitides with nervous system manifestations. Myasthenia gravis, another autoimmune condition, is discussed in Chapter 16. The paraneoplastic processes are so uncommon in the pediatric age group that they do not warrant discussion here.
EXPERIMENTAL MODELS FOR INFLAMMATORY DEMYELINATING DISEASE
Experimental allergic encephalomyelitis (EAE) and Theiler murine encephalomyelitis virus (TMEV) disease have been used as experimental animal models to study viral and autoimmune pathogenetic mechanisms in multiple sclerosis (MS) (1). The neuropathologic features and immunopathologic mechanisms responsible for inflammatory demyelination in EAE and TMEV are in many respects different. However, the models share certain similarities at the cellular and clinical levels insofar as they recall changes seen in human MS and acute disseminated encephalomyelitis (ADEM), for which they may serve as the two best experimental models (1,2,3).
Experimental Allergic Encephalomyelitis
Experimental allergic encephalomyelitis, also referred to as experimental autoimmune encephalomyelitis (4), has served for many decades as a useful animal model for the development and evolution of autoimmune diseases that affect the CNS. The EAE model was identified through efforts to elucidate the nature of a disseminated encephalomyelitis developing after human inoculation with Pasteur rabies vaccine (5). Because the vaccine was produced from virally infected neural tissue, animals were inoculated either with the vaccine or, for controls, uninfected neural tissue (5). However, during this exercise it was determined that some of the animals receiving uninfected neural tissue also developed encephalomyelitis (1,6).
Rivers and Schwentker were first to note that repeated injection of brain tissue into monkeys induced an inflammatory demyelinating encephalomyelitis (7).
Similar lesions have been produced consistently in other mammalian species; their appearance is enhanced by the addition of Freund’s adjuvant, which is a commonly used emulsion of water, oil, and killed acid-fast organisms added to the antigenic material. Its mode of action is unknown, but is believed to be a slow release of antigen inducing an inflammatory reaction that attracts mononuclear cells. In the original studies by Wolf and coworkers (8), 90% of monkeys developed EAE in 2 to 8 weeks after the first of an average of three weekly subcutaneous inoculations. The characteristic clinical features of this monophasic disease included paresis of the extremities, ataxia, nystagmus, and blindness. The disease was usually fatal, but some animals had mild symptoms that often subsided. A chronic and a relapsing disease marked by exacerbations and remissions reminiscent of the clinical picture of MS was produced subsequently in several animal species, including nonhuman primates (9,10).
Since then, the EAE model has been studied extensively owing to its clinical and histopathologic similarities to the human demyelinating diseases, especially ADEM (3) and multiple sclerosis (MS) (4,11,12).
Pathology
Postmortem neuropathologic examination of animals with EAE reveals multiple, multifocal areas of demyelination distributed throughout the neuraxis. Histologically, the demyelinating lesions exhibit a distinctive angiocentric
predilection and are accompanied by perivascular accumulations of mononuclear inflammatory cells (lymphocytes and monocytes) and perivascular microhemorrhages (13).
predilection and are accompanied by perivascular accumulations of mononuclear inflammatory cells (lymphocytes and monocytes) and perivascular microhemorrhages (13).
In addition, recent studies have demonstrated a range of axonal changes in EAE including axonal remodeling at multiple levels consistent with a highly plastic response of the motor system to inflammatory demyelinating insults (14). A comparative evaluation of acute axonal injury based on immunohistochemical reactivity for β-amyloid-precursor protein as a marker for dystrophic axons demonstrated similarities between patterns of axonal pathology in rats with myelin-oligodendrocyte glycoprotein (MOG)–induced chronic active EAE and human MS (15). The highest incidence of acute axonal injury was found during active demyelination, which was associated with axonal damage around demyelinating lesions and in the normal-appearing white matter of actively demyelinating cases. In addition, low but significant axonal injury was also observed in inactive demyelinated plaques. In contrast, no significant axonal damage was found in remyelinated shadow plaques (15).
Pathogenesis
EAE in the laboratory rat gives rise to an acute paralytic disease from which most animals recover spontaneously. The disease can be induced in genetically susceptible inbred Lewis and DA rats by direct immunization with myelin basic protein (MBP), encephalitogenic MBP peptides, or several other encephalitogenic proteins derived from myelin components, administered in complete Freund’s adjuvant (4). In addition, the disease can be adoptively transferred to syngeneic recipients with primed T cells [adoptive transfer of antimyelin-specific CD4+ T cells (16,17)] that have been reactivated in vitro with antigen (4).
Considerable variations in the susceptibility of various animals have been described, which are largely attributed to genotypic attributes, especially to the major histocompatibility complex (MHC) gene repertoire of the animal strain (18). That said, it should be noted that EAE is not a naturally occurring autoimmune disease except in genetically modified animal models such as in antimyelin-specific TCR/RAG-/- transgenic mice (19).
Studies of EAE in susceptible rats have provided many important insights into the interactions of T cells and accessory cells that culminate in the induction of the autoimmune response leading to inflammatory demyelination (4).
EAE immunopathogenesis revolves around predominantly cell-mediated autoimmune mechanisms. T cells are thought to play a pivotal role in initiating and perpetuating the myelinoclastic inflammatory process associated with EAE (5). The disease of the CNS is regarded as Th1 MHC class II restricted and CD4+ T cell mediated (5). EAE is mediated by CD4+ T cells that secrete cytokines (1,4,5). After stimulation and activation, T cells upregulate key adhesion molecules, facilitating their entry into the CNS (1). Moreover, Th1 proinflammatory cytokines secreted by CD4+ T lymphocytes augment the recruitment of mononuclear inflammatory cells in the CNS (4). In turn, cytotoxic T cells, activated monocytes/macrophages, and/or glial cells secrete cytotoxic factors leading to demyelination in conjunction with humoral responses in which B cells secrete antibodies against myelin antigens (1,4,5). Spontaneous remission is associated with CD4+ T cells that secrete transforming growth factor-beta (TGF-β) (4).
T cells recognizing antigenic determinants of myelin such as myelin basic protein (MBP), proteolipid protein (PLP), or myelin oligodendrocyte glycoprotein (MOG) are activated in the periphery and are subsequently recruited to the CNS through the action of chemokines to cause inflammation leading to neurologic signs including paralysis (20).
Normal individuals may harbor autoreactive CD4+ T cells, which, however, exist, as a rule, in a steady state of clonal deletion, T-cell anergy, and immunologic ignorance (5). Moreover, the peripheral immune system is endowed with a series of regulatory mechanisms that afford protection against both the generation of self-directed active immune responses and the initiation of autoimmune diseases (5). For example, CD4+ CD25+ regulatory T (Treg) cells can suppress such autoreactive T cells in EAE (21,22,23,24). Interestingly, alterations of such regulatory T cells were recently found in MS patients (25). In addition, the CNS is regarded as an “immunologically privileged” system, which is protected against peripheral immune responses by the tight endothelial junctions of the blood–brain barrier (BBB), the absence of dendritic cells in CNS parenchyma, and the presence of an immunosuppressive microenvironment (5). The latter is characterized by the secretion of anti-inflammatory cytokines and the expression of Fas ligand (CD154), which promotes T-cell apoptosis (5). However, this immunologic CNS homeostasis is perturbed and ultimately overcome when the CNS is exposed to inflammation, which causes opening of the BBB. The processes underlying T-cell priming and/or autoreactive T-cell dysregulation are unknown. T cell–mediated immune responses lead to the alteration of the BBB, facilitating the recruitment of other inflammatory cells, such as monocytes, as well as components of the humoral response (B cells and complement factors) in the CNS (26,27). In addition, cytokines produced by activated T cells in the lesions induce the activation of macrophages and local microglia effector cells, leading to the increase of their destructive activity, which is responsible for demyelination and tissue damage in MS (28).
Recent studies using genetically modified animals have elucidated two distinct clinical phenotypes of EAE in BALB interferon (IFN)-γ knockout mice immunized with
different residues of encephalitogenic peptides of MBP: (a) conventional disease, characterized by ascending weakness and paralysis, marked histologically by spinal cord inflammatory demyelination; and (b) a distinctive disease phenotype, characterized by uncontrolled axial rotation, involving demyelination of lateral medullary areas of brain (29). The type of disease is determined entirely by the inducing T cells, attesting to several divergent T cell–initiated effector pathways potentially involved in the pathogenesis of inflammatory demyelination (29).
different residues of encephalitogenic peptides of MBP: (a) conventional disease, characterized by ascending weakness and paralysis, marked histologically by spinal cord inflammatory demyelination; and (b) a distinctive disease phenotype, characterized by uncontrolled axial rotation, involving demyelination of lateral medullary areas of brain (29). The type of disease is determined entirely by the inducing T cells, attesting to several divergent T cell–initiated effector pathways potentially involved in the pathogenesis of inflammatory demyelination (29).
Although our current understanding of autoimmune inflammatory demyelinating disease of the CNS points to a role of regulatory T cells in EAE (21,22,23,30) and in MS patients (31), other regulatory mechanisms may also be involved in the immunopathogenesis of these disorders. These include the role of natural killer (NK) cells, natural killer T (NKT) cells, mast cells, B cells, and antibody responses in EAE and MS (5). The presumptive role played by B cells in the immunopathogenesis of MS is supported to some extent by the oligoclonal pattern of immunoglobulin production in the CSF in MS (32) and increased intrathecal immunoglobulin synthesis (33). As mentioned prevously, the disruption of the BBB that occurs in EAE and MS may facilitate the entry of B cells, antibodies, and complement into the CNS (5). B cells may then be activated or reactivated following T-cell interactions and become antibody-secreting cells (5). It is speculated that the demyelination observed during MS and EAE may represent an immunopathogenic synergy between autoimmune T and B cells through CD40–CD40L interactions, which underlie the development of humoral immunity (5,34,35). However, the significance of the presence of B cells and antibodies in the CNS is controversial and seems to be dependent on the experimental model used.
In summary, current understanding of the pathogenetic model of EAE and MS conforms to the following scheme of autoimmunity: Autoantigens are presented to T cells in a MHC context by antigen-presenting cells (APCs) such as dendritic cells in lymph nodes. Cooperation between T helper cells and B cells results in the recruitment of B-cell repertoires specific to autoantigens. Activated T cells are recruited to the CNS via chemoattraction and are reactivated by local or infiltrating APCs, resulting in the release of proinflammatory and cytotoxic mediators, leading to cellular injury. Vascular inflammation causes disruption of the BBB, which further facilitates the migration of T and B lymphocytes and monocytes and perpetuates recurrent immune-mediated injury to the CNS. The myelin sheath is damaged by several compounding mechanisms mediated by cytokines, complement, digestion of surface myelin antigens by activated macrophages, and direct damage by CD4+ and CD8+ T cells (5). Collectively, these pathways lead to cell death, including apoptosis, of oligodendrocytes and microglia (5) and axonal injury (15).
The usefulness of EAE as an experimental model for MS is likely to continue in the years to come. In particular, the reproduction of EAE in a nonhuman-primate model in the common marmoset (Callithrix jacchus), bridging the phylogenetic gap between rodents and humans, may further facilitate the elucidation of novel immunopathogenetic mechanisms and the development of more effective therapeutic strategies in MS and allied disorders (36).
Theiler Murine Encephalomyelitis Virus (TMEV) Infection
TMEV, DA strain, induces a biphasic disease in susceptible strains of mice (such as SJL), consisting of an early acute meningo-polioencephalomyelitis involving predominantly cerebral and spinal cord gray matter followed by a late chronic demyelinating disease with spinal cord white matter involvement akin to MS (2).
Pathology
In the early phase of infection, the disease is characterized by variously dense and multifocal inflammatory infiltrates involving cerebral and spinal cord gray matter (37). The inflammatory infiltrates are mononuclear and consist of lymphocytes (predominantly T cells) and monocytes/macrophages. Although there is lymphocytic infiltration of the leptomeninges and the cerebral cortex, most of the inflammation is present in the deep gray nuclei and mesotemporal region, especially in the thalamus, basal ganglia, and hippocampus (2). In the spinal cord, the inflammation is predominantly seen in the anterior horns, although infiltration of the leptomeninges is also present (2,38). During the acute early phase, there is sparing of the white matter throughout the neuraxis. The inflammatory infiltrates consist predominantly of perivascular CD3+ T cells and to lesser degree monocytes/macrophages. There is also evidence of incipient vasculitis in some small to medium-sized blood vessels (2).
Approximately 3 weeks after initial infection, there is infiltration of the spinal white matter by lymphocytes and monocytes/macrophages, which coincides with the onset of the chronic demyelinating phase of the disease marked by vacuolar change of the white matter, myelin loss, and aggregates of myelin-laden macrophages (2). Axonal swellings (spheroids) are detected in demyelinating lesions during the advanced stages of chronic disease. The demyelinating process is multifocal and involves all funiculi of the spinal cord (37). Demyelinating lesions are associated with perivascular and parenchymal inflammatory infiltrates comprised of CD4+ and CD8+ T cells, macrophages, and a few B cells, which are present predominantly in the spinal cord (37). There is widespread inflammatory infiltration of the spinal leptomeninges.
In the advanced chronic phase of the demyelinating disease (100 to 200 days postinfection) there is marked spinal cord atrophy without a concomitant increase in spinal cord demyelination (39). McGavern and colleagues (39) demonstrated a statistically significant loss of medium-sized and large myelinated axons only after the demyelinating phase of the disease is established. They speculate that following myelin denudation the naked axons are vulnerable to further inflammation and undergo dystrophic changes as a consequence of secondary damage (39). However, it is unclear whether the axonal damage in TMEV is the result of direct (inflammatory) injury or delayed wallerian or “dying-back” type of degeneration (39). On the other hand, Tsunoda and Fujinami (40) hypothesized that in TMEV, axonal injury is accompanied by oligodendrocyte apoptosis, which precedes demyelination, suggesting that an inciting axonal injury may be responsible for triggering demyelination.
The comparative neuropathology of TMEV-induced demyelinating disease in mice and MS in humans was discussed in the review article by Oleszak and colleagues (2).
Pathogenesis
Early acute disease is characterized by replication of the virus in gray matter (41,42). This phase of disease is associated with neuronophagia and inflammatory infiltrates in the cerebral cortical and deep gray matter as well as anterior horn cells of spinal cord (43). Infection with live TMEV is an essential component of TMEV demyelinating disease. TMEV-specific cellular and humoral immunity and apoptosis of infected cells eliminate virus from the gray matter of the CNS during the acute phase of TMEV disease (1,44). In particular, the virus is partially cleared from the CNS by CD3+ T cells, which undergo activation-induced apoptotic cell death, leading to resolution of the inflammatory response (44). Within 2 to 3 weeks the virus is partially cleared, and approximately 35 days postinfection susceptible mice develop late chronic demyelinating disease. In contrast to the acute phase, during the chronic phase, TMEV persistently infects glial cells and/or macrophages in the white matter (1,2,37,42,43,45,46). At the same time, recruitment of macrophages and T cells and generation of antibodies lead to inflammation and demyelination (1,2,44). Unlike the acute phase of TMEV infection, only very few mononuclear inflammatory cells (lymphocytes and macrophages) undergo apoptosis in the late phase of the disease, leading to the accumulation of these cells in the CNS, particularly in the spinal cord. It is believed that clonal expansions of T cells resistant to apoptotic clearance may play a pivotal role in the pathogenesis of demyelinating disease (2,44).
The fact that resistant strains of mice (such as C57BL/6) develop only early acute disease, are capable of clearing the virus completely, and do not develop delayed demyelination underscores the importance of genetic susceptibility, in the context of MHC genes, underlying the pathogenesis of TMEV-induced demyelinating disease (2,38,44). These strain-dependent, genetically determined immune responses to TMEV infection are also illustrated in the differential expression of proinflammatory cytokines in demyelinating disease–prone (SJL) versus disease-resistant (B6) mouse strains.
During early acute disease, there is a robust proinflammatory (Th1) cytokine response in the CNS of both TMEV-infected SJL and B6 mice, evidenced by the increased expression of polymerase chain reaction (PCR) transcripts for IFN-γ, interleukin (IL)-1, IL-6, IL-12p40, and tumor necrosis factor (TNF)-α (47). In the subacute phase of TMEV infection (8 days postinfection) TGF-β1 and TNF-α transcripts were present at significantly higher levels in the CNS of SJL susceptible mice as compared to resistant B6 mice (48). Concomitantly, TGF-β protein expression was demonstrated by immunohistochemical staining in leptomeningeal inflammatory cell infiltrates in brain sections of SJL mice but not in B6 mice. Chang and coworkers speculated that TGF-β may be responsible for the failure of SJL mice to mount an effective anti-TMEV circulating T-lymphocyte response (48). During late chronic demyelinating disease, there is an increase of proinflammatory Th1 cytokines in the CNS of disease-sensitive SJL mice as compared to disease-resistant B6 mice (48). Interestingly, increased expression of anti-inflammatory cytokine transcripts [IL-4, IL-5, and IL-10 (Th2 cytokines) and TGF-β] has been detected in the spinal cord of TMEV-infected SJL mice with chronic demyelinating disease as compared to the spinal cord of B6 mice. These anti-inflammatory cytokines may represent a compensatory mechanism of the host (disease prone SJL mice) in an attempt to downregulate proinflammatory cytokine responses in the CNS (48).
Thus, although oligodendrocytes and/or myelin may be damaged by a direct attack of cytotoxic T cells, other cells, including CD4+ T cells, activated macrophages, and microglia, may contribute to myelin destruction by the production of cytokines as well as reactive oxygen and reactive nitrogen species.
The role of inducible nitric oxide synthase (iNOS) has been investigated during early acute and late chronic TMEV-induced demyelinating disease. Both iNOS transcripts and protein have been detected in brains and spinal cords of TMEV-infected SJL mice during early acute disease, with significant decline during the chronic demyelinating phase of the disease (2,38,49). Immunohistochemically, iNOS has been detected in reactive astrocytes and in some monocytes during the acute phase of the disease but is distinctly absent in myelin-laden foamy macrophages in chronic demyelinating lesions (38). A similar trend has been observed in acute versus chronic human MS cases (50). It has been suggested that blockade of nitric oxide by treatment of TMEV-infected SJL mice with amino guanidine (AG), a specific nitric oxide inhibitor, results in delay of late chronic demyelinating disease (51). However,
this protective effect may depend on the temporal phase of the disease (early versus late), the type of cells expressing iNOS, and the time of administration of the nitric oxide inhibitor (49). It is speculated that nitric oxide production during early acute disease may be beneficial to the host through induction of apoptosis of infiltrating T cells and resolution of encephalitis, but its role in the pathogenesis of myelin or oligodendrocyte injury during late chronic demyelinating disease is unclear and may depend on other contributory factors (2,49).
this protective effect may depend on the temporal phase of the disease (early versus late), the type of cells expressing iNOS, and the time of administration of the nitric oxide inhibitor (49). It is speculated that nitric oxide production during early acute disease may be beneficial to the host through induction of apoptosis of infiltrating T cells and resolution of encephalitis, but its role in the pathogenesis of myelin or oligodendrocyte injury during late chronic demyelinating disease is unclear and may depend on other contributory factors (2,49).
TMEV-induced late chronic demyelinating disease is an excellent animal model for human MS (52), which, together with EAE, is likely to provide further critical insights into the pathogenesis and therapy of autoimmune demyelinating CNS disease in humans.
PRIMARY DEMYELINATING DISEASES OF THE CENTRAL NERVOUS SYSTEM
In Western countries with temperate climates, acute disseminating encephalomyelitis (ADEM), multiple sclerosis (MS), and optic neuritis are the three most frequently encountered primary demyelinating illnesses of the CNS (53). The concept of primary demyelination implies the destruction of the myelin sheets, oligodendrocytes, and Schwann cells with relative preservation of other components of the CNS (54). However, axonal injury is a common finding in demyelinating lesions, which correlates well with permanent functional deficits (55,56,57,58).
The demyelinating diseases of the CNS can be the consequence of (a) an immune-mediated inflammatory process resulting in destruction of the normally developed myelin (demyelinating or myelinoclastic diseases), (b) metabolic and genetic disorders of myelin metabolism, which embody abnormally formed myelin (dysmyelinating diseases) (3,59,60,61,62), or (c) a primary demyelinating process that occurs as a result of cerebral hypoxic-ischemic insults and certain forms of poisoning (54). Table 8.1 displays the classification of CNS myelin disorders.
ADEM is more common in children under the age of 12 years; MS is more common in adolescents and adults. Difficulty in distinguishing ADEM from the first bout of MS is among the most important reasons for the requirement of a second distinct episode occurring at least 1 month after the first for diagnosis of MS. It remains controversial as to whether “relapsing ADEM” should be distinguished from MS, but it appears likely that this distinction is ill defined in prepubertal children. Optic neuritis and the combination of optic neuritis and transverse myelitis (Devic disease) usually occur as manifestations of ADEM or MS, but pure transverse myelitis seems to be a distinctive nosologic entity, which may result from other type of illnesses (53).
An area of semiologic overlap exists between ADEM and Guillain-Barré syndrome (GBS). This area of overlap includes some or possibly all patients who manifest the clinical findings of Miller Fisher syndrome. It also includes the minority of ADEM cases that manifest diminished or absent muscle stretch reflexes in combination with weakness and sensory changes referable to peripheral nerve dysfunction. The designation of encephalomyelo-radiculo-neuropathy (EMRN) may be applied to cases exhibiting this overlap of central and peripheral demyelinating manifestations. Other considerably less common primary demyelinating conditions that may occur in children and are
often difficult to accurately classify include acute (Marburg type) MS, myelinoclastic diffuse sclerosis (Schilder disease), and concentric sclerosis (Baló disease). The disease encountered in infants younger than 2 years of age, who may experience a single bout of severe demyelination with edema, should be termed either acute MS or, perhaps more appropriately, severe ADEM (53).
often difficult to accurately classify include acute (Marburg type) MS, myelinoclastic diffuse sclerosis (Schilder disease), and concentric sclerosis (Baló disease). The disease encountered in infants younger than 2 years of age, who may experience a single bout of severe demyelination with edema, should be termed either acute MS or, perhaps more appropriately, severe ADEM (53).
TABLE 8.1 Classification of Central Nervous System Myelin Disorders | |
|---|---|
|
The etiology and pathogenesis of these various primary demyelinating illnesses are incompletely understood. Moreover, the degree of pathogenetic overlap among MS, ADEM, and other demyelinating diseases such as Devic disease and transverse myelitis is unknown. Both MS and ADEM are regarded as autoimmune diseases that involve cellular and humoral responses that are directed, at least in part, against myelin antigens. The onset of MS does not have a clear etiologic relationship to a preceding infection, and clinically discernible bouts of the disease are typically associated with detectable oligoclonal immunoglobulin production in the cerebrospinal fluid (CSF). ADEM appears in many cases to be provoked by an antecedent infectious illness and is accompanied by elevated CSF concentrations of immunoglobulins or immunoglobulin oligoclonality only in a minority of cases. Normal CSF immunoglobulin profiles are characteristic of recurrences of ADEM, as compared with a greater than 94% likelihood of abnormality in association with an MS recurrence (53).
A small minority of individuals who have experienced typical cases of ADEM in early childhood ultimately satisfy the clinical criteria for diagnosis of MS during adolescence, whereas others satisfy criteria for the diagnosis of MS with either relapsing-remitting or steadily progressive manifestations of primary central demyelination. There are no specific diagnostic tests or disease biomarkers to differentiate between ADEM and MS in the pediatric setting, and a number of other conditions must be excluded before entertaining either of these diagnoses. It may be particularly difficult to distinguish ADEM and related forms of inflammation from encephalitis. Indeed, some forms of encephalitis (such as those caused by herpes or measles viruses) may share overlapping clinicopathologic abnormalities with ADEM (53).
Multiple Sclerosis
Historical Aspects
MS is the principal immune-mediated demyelinating illness of humans (63,64). The pathologic lesions of MS were described by Cruveilhier and Carswell early in the nineteenth century. Frerichs was the first to make a clinical diagnosis of MS, in 1840. Charcot’s extensive studies of the clinical manifestations and natural history of MS resulted in diagnostic criteria for a coherent clinical entity designated disseminated sclerosis or sclérose en plaques disseminées, or Charcot disease (60). This condition was recognized at the outset exclusively among young adults. The occurrence of MS in children has been a topic for discussion for more than 50 years (53,60)
In 1922, Wechsler rejected most of the cases reported in pediatric populations but stated that “authentic cases of MS in children, in spite of their rarity, can occur (64a).” After that some isolated pediatric cases or small groups of affected children were reported (65). However, in that pre–computed tomography era, there was little evidence to identify the distinguishing characteristics of the condition in children. In 1948, Kabat and colleagues (66). reported increases in oligoclonal immunoglobulins in the cerebrospinal fluid of patients with MS, providing tangible evidence for an immunologically mediated inflammatory nature of the disease. In 1965, the diagnostic criteria of Schumacher and coworkers (67) established the age of debut of MS as being between 10 and 59 years, acknowledging that the condition does indeed occur below the age of 16 years.
A better understanding of the natural history of MS during the last 40 years has been made possible by important advances in neuroimmunology, molecular genetics, and biochemistry as well as the development of magnetic resonance imaging (MRI) techniques, which have allowed for an excellent and accurate anatomic visualization of the white matter in vivo. As a consequence, metabolic and infectious-inflammatory causes of demyelination have been delineated and the diagnosis of MS in children corroborated. Several retrospective studies were published in the 1980s and early 1990s, including those of adult patients with MS whose symptoms had been initiated during adolescence; the description of prepubertal patients was exceptional (68,69,70,71,72,73,74).
Today, it is generally accepted that MS can occur in children and even infants (75,76), although the distinctive characteristics of the disease at this age are not well established. It has been estimated that between 2.7% and 5.6% of patients with MS show symptoms attributable to the disease before 16 years of age (71,77,78,79,80,81,82,83). The frequency of MS beginning in early childhood has been calculated as 0.2% to 0.7% (71,84). According to these percentages, the corresponding prevalence of pediatric MS would be 1.35 to 2.5 per 100,000 people and that of the early infantile form would be 0.4 to 1.4 per 100,000 (85), although wider ranges. From 0.8 to 248 per 100,000 people, have been reported in Japan and Canada, respectively (77). Girls and women are at 1.5-fold to 2-fold greater risk than boys or men. Light-skinned individuals are at greater risk than more heavily pigmented individuals (64). However, MS can occur in black children, where it seems to have a rapidly progressive course (86). Residence in a northern climate before the age of 15 years confers an increased risk of developing the disease, although immigration to a southern climate at an early age substantially lowers the risk (87).
Pathogenesis
MS is clinicopathologically defined as a primary inflammatory demyelinating disease of the CNS. Although its etiology is unknown MS is widely regarded as an autoimmune disease involving predominantly abnormal cellular immune responses to putative (but not fully elucidated) autoantigens of central myelin (88). Over the years, two experimental animal models of MS have helped to better understand the pathogenetic mechanisms of this condition (see prior discussion of EAE and TMEV).
Epidemiologic data support the association of MS with hitherto unknown environmental factor(s) encountered during early childhood that after years of latency trigger the disease or contribute to its development. Three main groups of factors have been postulated in the immunopathogenesis of MS: (a) environmental factors, particularly the persistence of a viral infection, (b) immunologic factors involving autoimmune mechanisms with loss of tolerance to myelin antigens or molecular mimicry of viral antigens and myelin (or other host) proteins (2,88,89), and (c) genetic factors inducing a genetic predisposition to immunologic dysfunction.
Environmental Factors
Many investigators consider a viral infection to be the most likely environmental factor explaining the pathogenesis of MS. Indirect evidence supporting this theory comes from the particular distribution of the disease, with areas of high and low risk; serologic studies; isolation of viral proteins and genomic material from the brain of patients with MS; and different viral experimental models causing CNS lesions similar to MS pathology. Viruses probably related to MS include herpesvirus (in particular herpes human type 6), Epstein-Barr virus, paramyxovirus, and retrovirus. The recently discovered human endogenous retrovirus (HERV)-W family has an extracellular particle, named HSRV, that is associated to MS. Recent studies have shown that CSF levels of MSRV are related to the degree of CNS inflammation, and, even if this were an epiphenomenon, it could be used as a clinical prognostic marker of early MS (88).
Immunologic Factors
It is generally accepted that in addition to an early viral infection, there must be an autoimmune reaction that attacks some of the components of the myelin (2,88,90). Most patients exhibit T-cell reactivity to a number of myelin antigens, suggesting that by the time a patient develops clinical MS there has been epitope spreading with reactivity to multiple myelin epitopes (90). T cells that can react against myelin antigens are normally present in the immune system. These cells escaped thymic mechanisms of control such as clonal deletion.
A nontolerant, peripherally activated CD4+ T cell recognizes its autoantigen within the CNS parenchyma in the context of class II MHC molecules expressed by both local glial antigen-presenting cells (88) and dendritic cells (91), which commit T cells toward a Th1 phenotype. Activated Th1 cells cause myelin disruption and the release of new potential CNS autoantigens.
Secreted proinflammatory cytokines, such as IFN-γ and TNF-α, and chemokines recruit additional nonspecific inflammatory cells and specific antimyelin antibody–forming B cells, which exacerbate tissue injury (88,92,93). Finally, the apoptotic clearance of T cells and their conversion toward a Th2 phenotype modulates the outcome of the lesion (94). Additional cells are necessary for the development of typical MS lesions, such as the cytotoxic CD8+ cells, which show a more prominent clonal expansion within MS plaques and correlate better than CD4+ cells with the extent of acute axonal injury (95,96).
The cascade of inflammatory events that culminates in demyelination of axons depends on the peripheral activation of T lymphocytes (97). Interactions of lymphocytes with the vascular endothelium are required for lymphocyte trafficking into the CNS (2). Adhesion molecules play a critical role in this process and are pivotal in lymphocytes infiltrating the CNS through the BBB (98). One such molecule is the intercellular adhesion molecule-1 (ICAM-1), a glycoprotein that interacts with many α2-integrins such as lymphocyte function–associated antigen-1 (LFA-1) on T cells and CD11b/CD18 on monocytes (99,100). Other adhesion molecules mediating the interactions between lymphocytes and endothelial cells are very late antigen 4/vascular cell adhesion molecule-1 (VCAM-1), L-selectin (on lymphocytes) and E-selectin (on endothelial cells) (101,102).
Genetic Factors
Genetic factors have also been postulated to be contributors to the pathogenesis of MS, based on different studies. Prevalence rates for MS among first-degree relatives of individuals with MS are approximately 20-fold greater than those of other individuals from the same region (53). The risk of developing MS in the general population of 1 in 1,000 increases to 20 to 40 in 1,000 for first-degree relatives (87). In a pediatric series of 44 children with MS, 10 (23%) had a positive family history of MS in a first-degree relative (1 child), a second-degree relative (5 children), or their extended family (4 children). This is very similar to the 15% to 20% rate of a positive family history reported in an adult MS series (77). Identical twins have a 25% to 35% concordance rate for MS, as compared to 0.5% for offspring (possibly much higher for daughters of mothers with MS), 0.6% for parents, 1.2% for siblings, and 2% to 4% for dizygotic twins (87,103,104).
Guided by the considerations related to the presumptive immunopathogenesis of the disease, candidate gene searches have been focused on genes of the T cell–mediated immune response and of myelin proteins. Negative results have been found for the genes for T-cell receptor-α,
complement, chemokine receptors, interferons, other cytokine receptors, and the cytotoxic T-lymphocyte antigen 4 (CTL4), a gene located in the HLA class I region (88,103). There is a highly significant association of MS with HLA-DR2 and a weaker association with HLA-A3 and B7 in whites. Other candidates such as HLA class II have yielded positive results, especially when subtyped into HLA DRB1*1501, DQA*0102, and DQB1*0602 extended haplotype, although the estimated relative risk is only 2 to 4 (77,103,105). HLA-DR15 has been specifically associated with an earlier onset of MS in a large study of more than 900 patients (106).
complement, chemokine receptors, interferons, other cytokine receptors, and the cytotoxic T-lymphocyte antigen 4 (CTL4), a gene located in the HLA class I region (88,103). There is a highly significant association of MS with HLA-DR2 and a weaker association with HLA-A3 and B7 in whites. Other candidates such as HLA class II have yielded positive results, especially when subtyped into HLA DRB1*1501, DQA*0102, and DQB1*0602 extended haplotype, although the estimated relative risk is only 2 to 4 (77,103,105). HLA-DR15 has been specifically associated with an earlier onset of MS in a large study of more than 900 patients (106).
Recent analysis of markers of microsatellite polymorphisms in populations of different sizes and ethnicities identified chromosome regions of interest in MS susceptibility: chromosome 6 within the MHC and chromosomes 3q21–q24, 18p11, and 17q22–q24; a later meta-analysis, however, indicated the highest consensus evidence for linkage at 17p11 (107). All studies are concordant with the conclusion that HLA contributes, although modestly, to overall susceptibility and that a relatively large number of other MHC and non-HMC genes with individually small epistatic effects may be responsible for predisposition to MS. This implies a number of genes with interacting effects and suggests a polygenic inheritance of the disease. For example, the ApoE4 haplotype is also a genetic factor determining MS severity; these patients have a twofold higher likelihood of developing “black holes” on MRI and have an approximately fivefold greater rate of brain atrophy (108,109). It is probable that the expression of the implicated genes depends on environmental factors (88,103).
In summary, there is credence in the long-standing tenet of MS pathogenesis that the disease is produced by an environmental agent acting on a genetically susceptible individual in whom there are impaired immune responses.
Pathology
MS is defined as an inflammatory demyelinating disease of the CNS characterized by inflammation, demyelination, and axonal damage (58,95,110). The histologic hallmark of the disease, the MS plaque, is thought to be an end-point lesion. MS plaques reflect a continuum of immunologic activity encompassing inflammatory and secondary cellular changes in the affected brain. Morphologically, plaques are classified as acute (active), chronic active, chronic inactive, and “shadow” type (58,111). The histologic variability and the age of the plaques may be part of a temporospatial gradient of morphologic changes reflecting different immunologic mechanisms of cellular injury (95,111).
Plaque morphogenesis and evolution embody the interaction of immunologic and metabolic factors including the effects of cytotoxic T cells, antibodies, toxic metabolites derived from activated monocytes/macrophages, and metabolic derangements of oligodendrocytes (95,110,112,113,114,115). Our understanding of the pathogenesis of myelin destruction in MS is in keeping with the notion that plaques may represent a common morphologic end point of divergent immunologic pathways of myelin and axonal damage (114,116,117).
Gross Topography
The external appearance of the brain in patients with MS may be unremarkable. In the chronic cases, signs of atrophy with widening of sulci and slight enlargement of the ventricular system can be observed. When sectioning the brain, it is possible to identify firm, gray lesions on the surface of the brainstem, spinal cord, and optic nerves and multiple plaques of variable diameter in the white matter (118). Inflammatory demyelinating lesions are typically disseminated throughout the neuraxis, but are more frequently encountered in certain anatomic sites relating to recognizable patterns of neurologic semiology. Although the distribution of the plaques varies among patients, the preferential locations are: periventricular white matter, floor of the aqueduct and fourth ventricle (brain stem), cerebellar peduncles, cervical spinal cord, and optic nerves (Fig. 8.1).
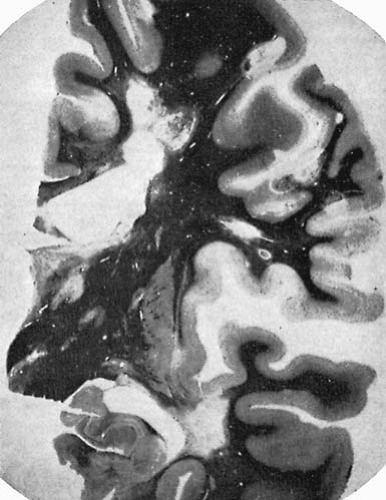 FIGURE 8.1. Multiple sclerosis. Disseminiated area of demyelination in white and gray matter of cerebral hemispheres. Myelin stain. (From Merritt HJ. Textbook of neurology, 5th ed. Philadelphia, Lea and Febiger, 1973. With permission.) |
Distinctive anatomic correlates are encountered in certain variants, such as the neuromyelitis optica or Devic type. Even though in the cerebral hemispheres the lesions have a periventricular predilection, an important proportion of lesions can be observed in other locations of central white matter and the gray–white matter junction (119,120,121). Lesions involving convolutional white matter typically spare the U fibers. There are instances in which white matter lesions extend into the contiguous cortex gray matter or deep gray nuclei (basal ganglia and thalami) (119,120). Subpial and intracortical plaque formation has been described and may represent an important correlate of neurologic disability (56). Exceptionally, large, spherical, tumor-like lesions are encountered (122), but such lesions are most commonly seen in conjunction with myelinoclastic diffuse sclerosis (Schilder disease) (see later discussion).
Histopathology
The MS plaques can be classified into four categories encompassing cellular changes that reflect disease activity or quiescence:
Acute (fresh) lesions: These are the early plaques characterized by perivascular inflammation (comprising predominantly lymphocytes and monocytes/macrophages), edema, myelin swelling, and activation of endothelial cells (58,95,110,111,119,120). Variable, often pronounced depletion of oligodendrocytes is present (58,123,124,125). Plasma cells are infrequent. There is apparent preservation of axons; however, incipient axonal injury may be present (57,58).
Chronic active lesions: These are full-blown plaques marked by active inflammation and demyelination, typically at their margins (i.e., interfaces with normal-appearing white matter). The following morphologic changes are present: perivascular lymphocytic infiltrates, ongoing myelin breakdown, myelin-laden macrophages, oligodendrocyte depletion, and reactive astrocytosis. The lymphocytic infiltrates may extend beyond the plaque margins (58,111,119,120,121). Ostensibly, there is sparing of axons, but variable degrees of axonal damage may be present (57,58,126).
Chronic inactive lesions: These are old, “burnt-out” plaques or quiescent lesions tantamount to glial scars (hence the classical use of the term sclerosis). They are sharply delimited from the adjacent, normally myelinated white matter. There is prominent astrocytic gliosis, loss of oligodendrocytes, and variable axonal damage ranging from dystrophic neurites to overt transection of axons and dendrites (58,111,119,120,121,126). Scant perivascular lymphocytic cuffs, monocytes, and/or plasma cells may be focally present. The walls of the blood vessels are sclerotic and hyalinized, pointing to antecedent inflammatory vascular injury. A thin rim of perivascular collagenous fibrosis may be present in long-standing lesions. The blood–brain barrier is disrupted (127).
Shadow plaques: These represent a variously sized and ill-defined zone of partially demyelinated or incompletely remyelinated tissue surrounding, and occasionally “overshadowing,” the principal plaque (128). Their occurrence in chronic MS is unpredictable, ranging from absent to frequent. There are no known clinical correlates, but “shadow plaques” may underlie a distinctive pathogenetic pathway (111).
Occasionally, the lesions are so severe that they evolve into cysts. It is classic to observe in the same patient lesions in different stages of progression.
Immunopathology
There are two integral components of MS lesions, perivascular inflammation and demyelination. It has long been hypothesized that inflammatory demyelination is the result of immune-mediated responses to myelin antigens in the myelin sheaths of axons and/or at the level of myelin-forming oligodendrocytes (95,110,119). Destruction of myelin and oligodendrocytes is not uniform in MS plaques (111,119). The morphogenesis of plaques is not fully understood, although circumstantial evidence points to early inflammatory damage of the BBB and infiltration by monocytes and lymphocytes, predominantly T cells (58,95,110,119). BBB disruption signals the onset of clinical symptoms, but a correlation between symptoms and inflammatory demyelination is not clear-cut.
Four distinct pathogenetic patterns of demyelination (I through IV) have been proposed (113). The first two focus on the concept that inflammation causes demyelination by direct and/or indirect mechanisms. Lymphocytes contribute to the pathologic process through cellular- and humoral-mediated immunologic responses (presumptive direct mechanisms) or by production of lymphokines and cytokines (indirect mechanisms). It follows then that patterns I and II exhibit remarkable similarities to either T cell–mediated or T cell plus antibody–mediated autoimmune encephalomyelitis (113). Monocytes/macrophages contribute to the demyelinating process in a twofold manner. First, through their traditional phagocytic role, cells of the monocyte/macrophage lineage, including hematogenously derived and activated resident microglia of the CNS, are potent effectors of axonal myelin and oligodendrocyte damage (58). Monocytes contribute to demyelination by way of production of cytokines, nitric oxide, and proteases and/or by directly targeting oligodendrocytes at the border of MS lesions (58,101,119). Activated CNS resident microglia play a role in the early stages of demyelination through cell-to-cell contact interactions with myelin internodes of the axons at the edges of active and chronic active MS lesions (58,119).
The other two patterns (III and IV) are consistent with a primary oligodendrocyte pathology (dystrophy) reminiscent of direct viral- or toxin-induced demyelination as opposed to bona fide autoimmune mechanisms (113). Over the years, various theories have been brought forward concerning the nature of oligodendrocyte damage in MS lesions. It is believed that this damage is incurred through a variety of immunologic mechanisms, including anti-MOG antibodies, cytokines produced by monocytes/macrophages and lymphocytes, T cell–mediated injury, immunoglobulins and components of activated complement, apoptosis, and a variety of other cytotoxic factors (58,114,115,129,130). In pattern III there is loss of myelin proteins in the distalmost (periaxonal) cell processes of oligodendrocytes, which is associated with apoptotic cell death (114,130,131). This mechanism has been previously defined as “distal” or “dying-back” oligodendrogliopathy (132) and is akin to oligodendroglial injury incurred during early hypoxic-ischemic demyelination of the white matter (130). In pattern IV there is cell death of oligodendrocytes in the white matter near active lesions (113). In this regard, it has been shown that activated monocytes/microglia expressing VCAM-1 selectively target oligodendrocytes at the border of MS lesions (101).
Axonal damage in the form of axonal swellings (dystrophic neurites) and transections has emerged as a major component of the disease (55,57,58,133,134). Axonal injury correlates with certain parameters of functional MR imaging (reduction of N-acetylaspartate) and certain patterns of neurologic disability in MS (15,114,126,135). Axonal pathology, evidenced by immunohistochemical staining for amyloid precursor protein (APP) in postmortem specimens, is more prominent in active MS lesions than in chronic inactive plaques (15).
Hypoxic-ischemic damage is an important but somewhat underrated aspect of MS neuropathology. Inflammatory damage of the vessel wall, endothelium, and BBB by T cells and monocyes (111,136,137,138) resembles the injury caused by angiocentric T-cell infiltrates in human immunodeficiency virus-1 (HIV-1)–associated CNS disease in children (139). The latter, compounded by edema and disturbance of the cerebral microcirculation, may impart damage to myelin, axons, and oligodendrocytes.
Disturbances in oxidative metabolism in MS reminiscent of hypoxia-ischemia may result from vascular factors (i.e., vascular inflammation) and/or the release of toxic metabolites associated with hypoxia-ischemia (130,140). Interestingly, overt ischemic damage has been demonstrated in severe cases of acute MS of the Marburg type, Baló concentric sclerosis, and neuromyelitis optica (141,142,143). It is hypothesized that certain active MS lesions may represent a form of “sublethal” hypoxic injury reminiscent of an ischemic white matter penumbra. Evidence of hypoxia-like metabolic tissue injury in MS due to the liberation of excitotoxins, reactive oxygen species, and nitric oxide lends further credence to this postulate (50,130,144,145,146).
In summary, current understanding of neuropathogenetic mechanisms in MS supports the hypothesis that white matter demyelination, axonal damage, dendritic transection, and apoptotic loss of neurons in the cerebral cortex contribute to neurologic dysfunction in MS patients (56).
Clinical Manifestations
Bauer and Hanefeld (147) classified MS according to the age at presentation: early infantile MS (EIMS), beginning between 1 and 5 years of age; delayed infantile or infantile MS (DIMS), beginning between 5.1 and 10 years; and juvenile MS (JMS), beginning between 10.1 and 16 years.
The clinical characteristics of 51 pediatric patients seen by one of us (S.N.T) (82,83) who fulfilled the MS diagnostic criteria of Poser and coworkers (148) are shown in Table 8.2. In 13 children disease was initiated before 5 years of age, and in another 13 it was initiated between 5 and 10 years. The youngest patient was an 18-month-old girl who has been followed for 16 years. So far, the youngest patient described in the literature is an 11-month-old infant (76).
The higher prevalence of infantile MS in girls was established in the series from Göttingen (147,149), which
included 20 children, with a ratio of 2.3:1. In our experience, this ratio was 1:3.3 in EIMS, 1:1.2 in DIMS, and 1.8:1 in JMS, the latter within the 1.5:1 to 1.9:1 ratio observed in adults (150). This finding suggests that hormonal changes related to puberty can interact with the immune and neuroendocrine systems and influence the course of some autoimmune diseases by modifying the humoral and cellular immune responses (151).
included 20 children, with a ratio of 2.3:1. In our experience, this ratio was 1:3.3 in EIMS, 1:1.2 in DIMS, and 1.8:1 in JMS, the latter within the 1.5:1 to 1.9:1 ratio observed in adults (150). This finding suggests that hormonal changes related to puberty can interact with the immune and neuroendocrine systems and influence the course of some autoimmune diseases by modifying the humoral and cellular immune responses (151).
TABLE 8.2 Clinical Data and Symptoms at the Time of the Initial Presentation of Multiple Sclerosis in 51 Children | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
The most frequent clinical presentation in children with EIMS and DIMS is an acute encephalopathy with multifocal deficits, more frequently acute hemiparesis with unilateral or bilateral pyramidal signs (81%). Other neurologic signs and symptoms present in 30% to 40% of patients include altered mental status, headache, vomiting, brainstem dysfunction, cerebellar ataxia, and meningeal signs. Most of the children recover from this dramatic picture after treatment with corticosteroids or may be left with mild residual deficits.
The patients with JMS, however, present isolated demyelinating syndromes, the most frequent one being the sensory hemisyndrome (64%), with or without associated motor findings, frequently without signs of acute diffuse encephalopathy. Acute loss of vision due to optical neuritis is observed more frequently as an initial symptom in children younger than 10 years of age (23%) than in those with JMS (16%). Seizures are infrequent (6% in our experience), and they present only in children less than 5 years of age, as part of the acute encephalopathy. Nevertheless, seizure frequency as high as 22% has been reported in early infantile forms of MS (80). Sensory findings, as indicated, are a frequent finding in these patients, and should suggest the diagnosis of MS. Paraparesis is often associated with abnormalities of posterior column function, which is often overlooked in adolescents because of an inadequate examination (53,64). Sometimes patients feel transient paroxysmal sensory phenomena such as sensation of a constricting truncal band or unexplained momentary exacerbation of sensory disturbance, associated or not with weakness, l’Hermitte sign (electric shock–like painful sensations spreading from the spine down into the extremities), and the Uhthoff phenomenon [transient appearance or worsening of neurologic dysfunction in association with exercise or exposure to hot ambient temperatures (atmospheric or while showering or bathing)] (53,64).
Children with MS more frequently present with a polysymptomatic form of the disease (43%) than a monosymptomatic one (36%), whereas the opposite is true for adults (35% and 65%, respectively) (77). Both children and adults commonly develop pyramidal signs, paresthesias, or myelopathy, but adults have a high incidence of brainstem and cerebellar signs and rarely manifest an acute encephalopathy (82,152,153).
Other symptoms that pediatric patients may experience as a consequence of MS include fatigue, spasticity, school difficulties, and emotional liability. Fatigue is described as “a subjective lack of physical or mental energy of sufficient severity as to interfere with the child’s ability to complete requisite school work, engage in extracurricular activities, or interact socially with peers” (77). Spasticity is one of the most common symptoms of MS; it hinders functional mobility, and is related to the course of the disease. Therefore it is more prominent in adults than children (154). Cognitive impairment has been demonstrated in pediatric MS patients (155,156). Adolescents with MS report difficulty with higher-order concepts and with organization of multiple tasks (77). The psychological impact of MS on the child or adolescent may be profound, although most children cope well with their diagnosis (77). More serious neuropsychiatric manifestations are seen in adults (157). Bowel and bladder dysfunction can also be an issue, although it is much less frequent in children than in adult patients (77). Epilepsy usually appears late in the course of the disease and, therefore, it is usually not seen in children with MS. Sometimes seizures may herald the onset of the condition or a relapse; they have a good prognosis (158,159,160).
Diagnosis
The diagnosis of MS in children is essentially clinical. It is supported by the neurologic examination, which reveals signs of white matter involvement with a defined temporal and spatial course, following the diagnostic criteria of Poser and colleagues (148). Additional studies, including MRI, CSF, and brain evoked potentials, complete the diagnostic process according to the new guidelines from the International Panel on the Diagnosis of MS (161) (Table 8.3)
Immunologic Studies
Immunologic abnormalities can be found both in the serum and the CSF. Dysfunction of cellular immunity is represented by an increase in the circulating CD4+ T-helper/inducer to CD8+ T-suppressor/cytotoxic cell ratio during MS relapses (53,162,163). Molecular markers of apoptosis (i.e., regulator CD95, caspases 8 and 10), and cytokine IL-10 and TNF-α in peripheral blood mononuclear cells correlate inversely with new MRI inflammatory activity, indicating that the CD95-dependent pathway plays a complex role in the regulation of survival of activated immune cells in MS (164).
CSF abnormalities in MS patients are characteristic, although neither specific nor pathognomomic. During the acute phase of demyelination there is lymphocytic pleocytosis of variable degree (30% to 70%), not generally exceeding 50 cells/mm3 (65,71,165,166). Dysfunction of the humoral immunity is a consistent finding in patients with MS and is represented by the almost universal presence in the CSF of (a) detectable oligoclonal immunoglobulins by electrophoresis, (b) elevated rates of synthesis and concentrations
in the CSF of intrathecally generated immunoglobulin G (IgG) and IgM with varied or unknown epitopic specificity, and (c) increased levels of immunoglobulin components such as kappa chains (53,65,71,165,166,167,168,169). The finding of oligoclonal bands (OCB) of IgG present in the CSF and absent in blood has been described in 65% to 95% of adult patients with MS (33,166,170,171). The reported frequency of OCB in children with MS is variable, which is probably a reflection of the different methodologies used (172). With the isoelectric approach followed by inmunofixation, Tenembaum and colleagues (82,83) detected positive CSF OCB in 27 of 51 (53%) children with MS. The detection rates for the three clinical forms, EIMS, DIMS, and JMS, were, respectively, 46%, 38%, and 64%. Recently, the serum analysis of antimyelin antibodies, notably anti-MOG and anti-MBP, has been shown to be relatively effective in predicting the progression of isolated demyelinating syndromes to full blown MS (173). Rejdak and collaborators (174) reported increased CSF levels of nitric oxide metabolites (nitrite and nitrate levels) in adult MS patients, which correlated with clinical and MRI progression of the disease over a 3-year follow-up. Sueoka and colleagues (175) found selective CSF synthesis of antibodies against ribonucleoprotein B1 in adults with relapsing-remitting MS, suggesting that they could be a disease marker for MS.
in the CSF of intrathecally generated immunoglobulin G (IgG) and IgM with varied or unknown epitopic specificity, and (c) increased levels of immunoglobulin components such as kappa chains (53,65,71,165,166,167,168,169). The finding of oligoclonal bands (OCB) of IgG present in the CSF and absent in blood has been described in 65% to 95% of adult patients with MS (33,166,170,171). The reported frequency of OCB in children with MS is variable, which is probably a reflection of the different methodologies used (172). With the isoelectric approach followed by inmunofixation, Tenembaum and colleagues (82,83) detected positive CSF OCB in 27 of 51 (53%) children with MS. The detection rates for the three clinical forms, EIMS, DIMS, and JMS, were, respectively, 46%, 38%, and 64%. Recently, the serum analysis of antimyelin antibodies, notably anti-MOG and anti-MBP, has been shown to be relatively effective in predicting the progression of isolated demyelinating syndromes to full blown MS (173). Rejdak and collaborators (174) reported increased CSF levels of nitric oxide metabolites (nitrite and nitrate levels) in adult MS patients, which correlated with clinical and MRI progression of the disease over a 3-year follow-up. Sueoka and colleagues (175) found selective CSF synthesis of antibodies against ribonucleoprotein B1 in adults with relapsing-remitting MS, suggesting that they could be a disease marker for MS.
TABLE 8.3 Diagnostic Criteria of Multiple Sclerosis | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||
Neurophysiology
Neurophysiologic studies are not specific. The electroencephalogram (EEG) can show changes corresponding to the epilepsy diagnosed in some patients (148,158,160). Aphasic status epilepticus, epilepsia partialis continua, and periodic lateralized epileptiform discharges have been reported (176,177,178). Prolonged treatment with corticosteroids induces changes of the sleep EEG in MS patients similar to the changes observed in patients with an acute depressive episode (179). Evoked potentials (EPs) provide information about dissemination of demyelinating disease within the CNS (77). Visual and somatosensory EPs can demonstrate a second lesion (180). Although their usefulness in pediatric MS has yet to be formally evaluated, abnormalities in visual and somatosensory EPs have been demonstrated (68) and are likely to be of similar diagnostic significance as in adult MS patients (181,182,183).
Anatomic Neuroimaging
Brain and spinal cord MRI are the neuroimaging modalities of choice for evaluating children with demyelinating disorders in general and MS in particular. The typical MRI lesions described in adult patients with relapsing-remitting MS are round or oval plaques, bright or hyperintense on T2-weighted, proton density, and fluid-attenuated inversion recovery (FLAIR) images. They are
variable in number and distributed in the white matter of the centrum semiovale, adjacent to the ventricles, and in the corpus callosum, brainstem, cerebellum, optic nerves, and spinal cord (118,181). These lesions are perpendicular to the lateral ventricles, are usually small (less than 5 mm), and show an incomplete, nonuniform enhancement with gadolinium. In the experience of one of us (S.N.T.) the cerebral images of patients with juvenile MS are not different from this classic description at the time of the initial presentation or during relapses (82,83) (Fig. 8.2A).
variable in number and distributed in the white matter of the centrum semiovale, adjacent to the ventricles, and in the corpus callosum, brainstem, cerebellum, optic nerves, and spinal cord (118,181). These lesions are perpendicular to the lateral ventricles, are usually small (less than 5 mm), and show an incomplete, nonuniform enhancement with gadolinium. In the experience of one of us (S.N.T.) the cerebral images of patients with juvenile MS are not different from this classic description at the time of the initial presentation or during relapses (82,83) (Fig. 8.2A).
In contrast, brain MRIs performed during the initial demyelinating episode in children younger than 10 years of age show large multifocal demyelinating plaques with a tendency to coalesce (Fig. 8.2B) in 80% of cases (83), a finding that is somewhat underrated (72). These lesions can have a tumefactive (tumor-like) appearance with variable mass effect. Usually these can be differentiated from tumors by the lesser amount of edema around the lesions frequently (but not invariably) in association with other typical, smaller lesions (184) (Fig. 8.2C). That said, there are reported cases of infantile MS with a solitary plaque associated with perilesional edema (185). The enhancement with gadolinium can be helpful for establishing this difference because the demyelinating plaque usually shows an enhancement pattern of incomplete “hoop” or “open ring” (186). Up to 15% of patients with juvenile MS may have tumefactive lesions on the MRI at the time of the initial attack. The presence of “black holes” on unenhanced T1-weighted images and signs of brain atrophy, like widened subarachnoid spaces, ventriculomegaly, and thinning of the corpus callosum, have not been frequently described in the pediatric literature, but these findings are clearly seen in children with secondarily progressive clinical forms of MS (82).
The MRI images observed in children with an initial attack of EIMS or DIMS recall the images of patients with ADEM (187,188). Characteristic lesions are large and disseminated in the subcortical white matter, but also in the cortex and deep gray nuclei, without the distribution and morphology observed in juvenile and adult MS. As a rule, only if subsequent studies at the time of clinical follow-up or relapses show new lesions that enhance with gadolinium can the diagnosis of MS can confirmed (161,189,190). With this in mind, it should be recognized that ADEM can be accompanied usually by one or several episodes of relapse (biphasic or multiphasic ADEM) (161,189,190), but successive MRIs will reveal active lesions only in the context of a concomitant clinical attack; in other words, “subclinical” or “silent” lesions exhibiting active MRI changes (that are typical of MS) are not seen in ADEM (188,191).
According to the new guidelines from the International Panel on the Diagnosis of MS, (161,192), in certain clinical situations, MRI lesions must fulfill the diagnostic criteria of space dissemination, which include three of the following four: (a) one gadolinium-enhancing lesion or nine T2 hyperintense lesions if there is no gadolinium enhancement, (b) at least one infratentorial lesion, (c) at least one juxtacortical lesion, and (d) at least three periventricular lesions. One spinal cord lesion can be substituted for one brain lesion (161). However, these criteria may not apply as well to the pediatric MS population. Children with MS appear to have fewer white matter lesions at the time of their MS diagnosis than do newly diagnosed adults. Moreover, because myelinogenesis is incomplete in childhood, this may influence lesion appearance, size, and distribution within the CNS. Further studies are necessary to develop MRI diagnostic criteria that are validated in the pediatric MS population (193).
MRI and MR spectroscopy (MRS) in patients with MS for less than 5 years shows brain atrophy and loss of axonal integrity. Although the exact mechanisms underlying CNS atrophy in MS patients are largely unknown, evidence exists that atrophy may be secondary to the repeated effects of inflammatory demyelination, axonal injury, including dystrophic changes and frank axonal transection, wallerian degeneration, and neuronal loss (56,194,195).
Functional Neuroimaging
Although routine MRI is the mainstay of diagnosis of MS, there is increasing interest in using quantitative MRI methods to better understand pathology in gray matter and normal-appearing white matter. Magnetization-transfer MRI and diffusion-weighted MRI are techniques that provide additional useful information about the process of demyelination and remyelination in MS (196). In addition, functional neuroimaging studies like MRS, functional MRI (fMRI), single photon emission computed tomography (SPECT), and positron emission tomography (PET) allow a better study of the CNS dysfunction secondary to the pathologic changes of the disease (194,196).
MRS of the cerebral demyelinating plaques in children shows a spectrum of alterations with reduction of N-acetylaspartate (NAA) and creatine and increase of choline and myo-inositol compared with age-specific controls (71,197). MRS in adults with MS may also show elevation of lactate in the acute demyelinating plaque, but this finding has not been reported in children (74). The white matter adjacent to the plaques that has a normal appearance on MRI has a normal metabolic pattern on MRS, but the adjacent gray matter usually shows a decrease in NAA (197). These data are similar to the findings in adult patients, and are the consequence of neuronal, including axonal, injury in addition to myelin damage that occurs early as a result of repeated inflammatory demyelinating insults (194,196).
fMRI is being used in clinical research to study the neuronal mechanisms that underlie CNS function and to define abnormal patterns of brain activation that arise from
disease (196,198). A changed pattern of cortical activation, mainly characterized by increased activation of the contralateral primary sensorimotor cortex, has been found in MS patients with clinically isolated syndromes when they do a simple motor task (199). A strong correlation has also been found between the extent of activation of the contralateral primary sensory motor cortex and the reduction of the whole-brain NAA concentration, which suggests that functional cortical reorganization might contribute to the maintenance of normal cortical function in the early stages of MS. Increased activation of several sensorimotor areas, mainly in the cerebral hemisphere ipsilateral to the extremity used to do the task, has also been reported in patients with early MS and preceded by an episode of hemiparesis (200). In patients with similar characteristics but with optic neuritis as their first clinical manifestation, sensorimotor areas mainly located in the contralateral cerebral hemisphere were recruited (201). Abnormal brain activation of the prefrontal/frontal lobe has been demonstrated in MS patients with fMRI during specific tasks; the dysfunction normalizes with rivastigmine, a central cholinesterase inhibitor (202).
disease (196,198). A changed pattern of cortical activation, mainly characterized by increased activation of the contralateral primary sensorimotor cortex, has been found in MS patients with clinically isolated syndromes when they do a simple motor task (199). A strong correlation has also been found between the extent of activation of the contralateral primary sensory motor cortex and the reduction of the whole-brain NAA concentration, which suggests that functional cortical reorganization might contribute to the maintenance of normal cortical function in the early stages of MS. Increased activation of several sensorimotor areas, mainly in the cerebral hemisphere ipsilateral to the extremity used to do the task, has also been reported in patients with early MS and preceded by an episode of hemiparesis (200). In patients with similar characteristics but with optic neuritis as their first clinical manifestation, sensorimotor areas mainly located in the contralateral cerebral hemisphere were recruited (201). Abnormal brain activation of the prefrontal/frontal lobe has been demonstrated in MS patients with fMRI during specific tasks; the dysfunction normalizes with rivastigmine, a central cholinesterase inhibitor (202).
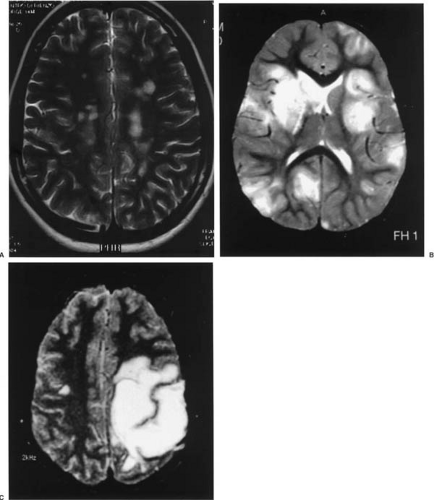 FIGURE 8.2. A: Juvenile MS. Axial T2-weighted magnetic resonance imaging (MRI) showing small, hyperintense lesions in the periventricular white matter. B: Early infantile multiple sclerosis (MS). Axial T2-weighted MRI with numerous bilateral, predominantly subcortical demyelinating lesions. Right thalamic lesion shows expansive effect with partial ventricular collapse. C: Juvenile MS. Axial T2-weighted MRI, with extensive tumefactive lesion in the left frontoparietal white matter. A “satellite” small lesion can be observed in the contralateral white matter. |
New functional MRI techniques are being developed to study in vivo CNS diseases at a molecular level (194). Experimental studies with Theiler murine encephalomyelitis virus have investigated MRI techniques using antibodies linked to superparamagnetic particles directed against immune-specific immune determinants. This allows selective imaging of CD4+ T cells, CD8+ T cells, and Mac1+ cells in the CNS (203). Being able to monitor dynamically the activity of specific classes of inflammatory cells in MS will provide an important way of understanding the evolution of pathology and the effects of interventions (194).
In a study of 17 MS patients, 99mTc-D,L-hexamethylpropylene amine oxime (99m Tc-HmPAO) SPECT showed reduced ratios of regional to whole-brain activity in the frontal lobes and left temporal lobe (204). A relationship was found between left temporal lobe abnormality and deficit in verbal fluency and verbal memory. SPECT is also useful in evaluating MS patients with depressive disorders (205) and in establishing the differential diagnosis of tumor-like lesions (206).
PET studies have demonstrated that global and regional cortical glucose metabolism is significantly reduced in MS patients compared with normal controls. Such a decrease correlates with number of relapses, total lesion area on MRI, and cognitive dysfunction, indicating that MRI white lesion burden causes deterioration of cortical cerebral neural function (207,208,209). Hypometabolism is widespread, including the cerebral cortex (frontal, parietal, occipital), supratentorial white matter (parietal), and infratentorial structures (pons) (208,210). Using a radioligand for the peripheral benzodiazepine receptor, PET has allowed the visualization of microglia and its involvement in the inflammatory processes causing MS (211).
Differential Diagnosis
The differential diagnosis of MS is broad. Although the clinical signs and symptoms, the MRI changes, and the CSF findings are characteristic of MS, they are not specific and can be found in other inflammatory or infectious conditions, as given in Table 8.4.
TABLE 8.4 Differential Diagnoses of Multiple Sclerosis | ||
|---|---|---|
|
Treatment
The treatment of MS in children, as in adults, must include the suppression of the inflammatory immune responses during relapses and the amelioration of the associated inter-relapse symptoms (i.e., fatigue, spasticity, urinary tract infections) (77).
Initial treatment consists of the administration of corticosteroids, either orally or intravenously (212). The use of an intravenous (IV) pulse of methylprednisolone is indicated in severe attacks, characterized by marked involvement of mental status, optic nerves, or spinal cord, or in cases with tumefactive lesions on MRI (213). The recommended dose is 30 mg/kg per day to 1 g/day, administered as a 1-hour infusion on 3 to 5 consecutive days, followed by oral administration of methylprednisone, 1 mg/kg per day in the morning during the next 10 days, followed by tapering over a 3-week period. Administration of IV methylprednisolone hastens recovery from acute exacerbations of the disease (77). Recent studies have shown that methylprednisolone suppresses the expression of genes associated with T-cell differentiation and activation, which may contribute to its beneficial effect in relapses of MS (214). Treatment with corticosteroids requires careful monitoring of blood pressure, urine glucose, and serum potassium and administration of gastric protection.
Relapses that are not as severe can be treated with oral methylprednisone at a dose of 1 to 2 mg/kg per day for 10 to 15 days. By and large, very mild exacerbations manifested only by sensory symptoms like paresthesias do not require treatment. Chronic administration of corticosteroids is not indicated because it has been shown that they do not modify the natural history of the disease in adult patients and can cause serious side effects in children (213).
Some children who do not respond to IV corticosteroids may benefit from IV immunoglobulin (IVIG) (77). The recommended dose is 2 mg/kg in divided doses over 2 to 5 days. There are no studies of IVIG efficacy in pediatric MS. Its efficacy in adults was recently reviewed in a meta-analysis, which suggested a potential role for IVIG in patients with high relapse frequency (215). Similarly, IVIG treatment for the first year from onset of the first neurologic event suggestive of MS significantly lowers the incidence of a second attack and reduces disease activity as measured by MRI (216). In a single-blind study of adolescents and adults, IVIG showed a similar efficacy to INF-β1a in decreasing the relapse rate in relapsing-remitting MS (217). In contrast, IVIG did not show any clinical benefit in a group of adult patients with secondary progressive MS (218).
The efficacy and long-term benefits of other immunosuppressive agents in MS are unclear. In this regard, there are only a few trials in adults and none in children. Azathioprine has been used alone and in combination with IVIG (217) or IFN-β1b (219). Although this immunosuppressive agent appears to reduce the relapse rate in MS patients, its effect on disease progression and neurologic disability has not been established (220). Methotrexate may alter the course of disease favorably in patients with progressive MS, but the evidence is tenuous (215).
Cyclophosphamide has been used in adults with variable success. It appears to be more efficient in the early stage of progressive MS independent of age, relapses, or neurologic disability scale (221). The preliminary analysis of a randomized, single-blind, parallel-group, multicenter trial in MS patients of pulse cyclophosphamide demonstrated it to be a therapeutic option as rescue therapy for patients who are IFN nonresponders (222). In addition to having a general immunosuppressant effect, cyclophosphamide has selective immunosuppressant effects in MS by suppressing IL-12 Th1-type responses and enhancing Th2/Th3 responses (IL-4, IL-10) (223). Cyclophosphamide has been used in a few children with very aggressive MS after failed IVIG and IFN therapies. Potential risks of immunosuppression, malignancy, and infertility should be carefully weighted against potential benefits and be thoroughly discussed with the patient and family (77).
Mitoxantrone is another potent immunosuppressive agent used to treat MS in adults. In a study of 94 patients followed for 3 years, it was effective in improving or stabilizing the course of the disease, particularly in patients with a low rate of relapses (224).
Plasmapheresis has shown efficacy in some studies of adult MS patients (225,226,227). However, its efficacy in the treatment of MS is controversial (228).
The usefulness of immunomodulatory treatments, which have been demonstrated to improve the natural history of MS in adults, such as IFNs (229,230,231,232) and glatiramer acetate (copolymer 1, or Copaxone) (233), has not been investigated with controlled studies in children, although there is evidence of success in isolated cases (77). IFN-β1b (Betaseron, Betaferon) is tolerated well and has been shown to produce clinical benefit in children with MS (234,235,236). Tenembaum and colleagues (213,237) reported their experience treating 31 children with IFN-β1a (Avonex) 30 μg once a week, IFN-β1a (Rebif) 22 μg three times a week, IFN-β1b (Betaseron), 250 μg every other day, and glatiramer acetate (Copaxone) 20 mg daily. The tolerance to the four treatments was similar to that reported in adult patients. Children showed flulike symptoms in 61% of cases, reactions at the site of injection in 39%, migraines in 29%, and systemic transient reaction in 6%; these symptoms gradually decreased until their disappearance in a few months. The doses were titrated according to age, weight, and clinical response. Analysis of the efficacy showed a significant reduction of the rates of relapse in 94% of patients with relapsing-remitting MS and in 33% of patients with secondary progressive MS.
Preliminary clinical trials suggest that combination therapy in MS does not increase the side effects of approved monotherapy; its efficacy over monotherapy should therefore be tested (238).
Natalizumab (Tysabri) is an anti–α4-integrin monoclonal antibody that blocks α4β1-integrin–mediated leukocyte migration. The latter binds to vascular cell adhesion molecule-1, which is expressed at high levels in the blood vessels in the CNS during MS exacerbations (239). An open-label studied of 38 patients with relapsing-remitting MS stable on treatment with IFN-β1a demonstrated the safety of this combination (240), and a large multicenter study provided efficacy results sufficient for the U.S. Food and Drug Administration to approve Tysabri in 2004 as an IV formulation for the treatment of multiple sclerosis (and Crohn disease) in adults.
Other therapeutic approaches that are being investigated and may provide new therapeutic alternatives for MS in the future are (a) selective immunosuppression against T-cell homing (241); (b) immunomodulation using new drugs like statins (242) or targeting new mechanisms like chemokine receptors or dendritic cells that inhibit T cells (243,244), neuroprotective agents like erythropoietin (245), or T-cell migration per se with antimetalloproteinases (246) or antioxidants (247); (c) interventions aimed at iron and iron-mediated production of free radicals (248); (d) T-cell vaccination (249); (e) stem cell transplantation (250); and (f) gene therapy (251)
Symptomatic treatment is paramount to improving the quality of life of patients with MS (252). Modafinil and amantadine are effective in treating fatigue (253). Exercise and yoga (52) have also demonstrated a beneficial effect. Depression responds well to pharmacotherapy (253a). Spasticity (154) and epilepsy (158,159,160) should be treated with the appropriate specific medications.
A multidisciplinary approach to the care of these children and their families through assessment, support, education, advocacy, referral, and coordination of care is ideal for the best control of the disease and the best quality of life (254).
Prognosis
In adult patients, the clinical course of the disease is highly variable. Relapsing-remitting MS is the most frequent form of progression (80%), followed by secondary progressive MS after an initial period of frequent bouts (26%) and primary progressive MS (6% to14%).
There is a scarcity of information about the clinical course and prognosis of MS in children. In a study of 296 children having a first episode of acute CNS inflammatory demyelination, the rate of a second attack was higher in patients with age at onset older than 10 years, MS-suggestive initial MRI, or optic nerve lesion and lower in patients with myelitis or mental status change (255). In the series of one of us (S.N.T.) (83) the clinical course of a group of children with an established diagnosis of MS was relapsing-remitting (73%) or secondary progressive (25%). No patient developed primary progressive MS. Other authors have published similar findings (78,256)
The natural history of MS in adults shows that in the long term the frequency of disease exacerbations decreases spontaneously (257). In the experience of Tenembaum and colleagues (83) the mean number of bouts per year was 2, 2.3, and 2.2 for EIMS, DIMS, and JMS, respectively, during the first year of disease. These figures decreased to 0.9, 1.4, and 1.5, respectively, at the time of 4 to 8 years follow-up. These findings indicate that children with MS have a tendency to undergo more relapses of demyelination than adults, although they also show a spontaneous reduction of the frequency of relapses with time.
Relapses can be provoked by acute febrile viral illnesses. Bacterial infections, typically those involving the urinary tract of girls or women with MS, may provoke a relapse, but it is more likely that they worsen the degree of already existing disease activity. There is no proof that live-virus vaccine or influenza vaccine provokes relapses. Other factors that may be associated with a relapse include stress, physical trauma, surgery, and spinal anesthesia (53).
As the frequency of relapses increases, recovery is hampered and deficits may become cumulative. As a rule, the longer the bout, the greater is the likelihood that recovery will be incomplete (53). The evaluation of neurologic disability is usually performed utilizing the Expanded Disability Status Scale (EDSS) of Kurztke (258,259). However, this method, which is widely used in adults, shows clear limitations in children. In the series of Tenembaum and colleagues (83), after a mean follow up of 6.6 years, 70% of children with MS had an EDSS score of 3.5, that is, they were ambulatory without assistance; 9% had scores between 4 and 4.5, indicating moderate disability with limitations, but still ambulatory; 15% had scores ranging from 6 to 9.5, meaning that they ambulated with assistance or were wheelchair bound. These data demonstrate that MS in children, both the infantile and juvenile variants, cannot be considered a benign disease because its clinical course can be aggressive and cause significant physical handicap.
Children in the series of Mikaeloff and coworkers (255) were followed up for a mean of 3 years. Ninety percent had no or minor disability. Occurrence of severe disability was associated with a polysymptomatic onset, sequelae after the first attack, further relapses, and progressive MS.
Variants of Multiple Sclerosis
Three variants of MS that have attracted considerable attention among neurologists are Schilder disease (myelinoclastic diffuse cerebral sclerosis), Devic disease (neuromyelitis optica), and Baló disease (encephalitis periaxialis concentrica).
Schilder Disease (Myelinoclastic Diffuse Cerebral Sclerosis, Encephalitis Periaxialis Diffusa)
Myelinoclastic diffuse sclerosis (MDS) is a rare demyelinating disorder of the CNS that affects mainly children but also adults (3,261,262). The disease was originally described by Paul Schilder in 1912 under the name encephalitis periaxialis diffusa (263). Schilder subsequently used the same designation to describe two additional cases in 1913 (264) and 1924 (265). However, in the intervening years, the term Schilder disease was applied to a variety of fundamentally disparate CNS diseases affecting myelin. These include MDS (the type in Schilder’s original 1912 case), which exemplifies the nosologic position of this disease entity as a variant of MS; leukodystrophies (Schilder’s 1913 case); instances of postmeasles subacute sclerosing panencephalitis (SSPE) (Schilder’s 1924 case); and atypical cases of ADEM (3,266). This nosologic confusion was eventually clarified in 1956 by Poser and von Bogaert (267), who after analyzing the cases compiled by Bouman in his extensive monograph of the disease (268), found that more than one-half of cases described as Schilder disease or MDS were in fact leukodystrophies, ischemic demyelinations attributed to hypoxic-ischemic encephalopathies, or SSPE. Subsequently, Poser (261) showed that in many cases of MDS, typically scattered lesions of MS also existed in addition to the larger areas of demyelination, attesting to the fact that Schilder’s 1912 case was indeed another form of MS, more commonly seen in the young (3). In recent years, additional confusion has been introduced by indiscriminately applying the diagnosis of Schilder disease to cases characterized by large areas of increased signal intensity on T2-weighted MRIs that actually represent instances of ADEM (269).
Currently, Schilder disease (or MDS) is considered to be an inflammatory demyelinating disease of unknown etiology, which constitutes a distinctive clinicopathologic variant of MS affecting the white matter of the cerebrum and the entire neuraxis (119,262,270). The disease is distinguished by two features: First, there is a propensity to produce one or more fairly symmetric, large, often spherical and tumefactive demyelinating lesions in the centrum ovale and central white matter with sparing of the subcortical U fibers (271). Second, there is an exquisite sensitivity to treatment with corticosteroids (272).
Pathology
On anatomic examination of the sectioned brain, the most striking alteration is the grossly discernible demyelination of the central hemispheric white matter. Typically, the plaques are fewer and larger than in classical MS and their deep location with sparing of the U fibers at the gray–white junction serves as a topographic distinguishing feature from ADEM. Remarkably, even in the most severely affected white matter with near total loss of myelin, a narrow band of subcortical, convolutional white matter is usually spared (Fig. 8.3). The lesions are most common in the frontal and posterior parietal/occipital lobes, but can involve any part of the cerebral hemispheres, brain stem, cerebellum, and spinal cord (119).
 FIGURE 8.3. Diffuse cerebral sclerosis (Schilder disease). Myelin preparation of the frontal lobe demonstrating demyelination. The arcuate fibers are characteristically spared. (Courtesy of the late Dr. D. B. Clark, University of Kentucky, Lexington. KY.) |
Histopathologically, the lesions are indistinguishable from active inflammatory demyelinating lesions of MS (262) or ADEM, although there is a distinctive tendency for central necrosis and cavitation especially within larger lesions (119). Histologic sections of the gross specimen confirm the presence of an inflammatory demyelinating process. As a rule in autopsy cases, lesions are of the same age. When they are extensive, multifocal, and confluent they are typically characterized by massive accumulations of foamy or myelin-laden macrophages imperceptibly merging with hypertrophic reactive astrocytes. The former are highlighted by immunohistochemistry using macrophage markers such HAM-56 and CD68, whereas the latter are delineated by robust staining for glial fibrillary acidic protein (GFAP) (50). Focal perivascular mononuclear, lymphocytic-monocytic infiltrates are present.
The pathogenesis of MDS is unknown, but it likely involves similar immunopathogenetic mechanisms to those of MS. As indicated in the discussion of MS pathogenesis, an inflammatory-type oxidative stress injury marked by increased expression of iNOS and nitrotyrosine has also been reported in cases of MDS (50).
Clinical Manifestations
The illness tends to present in children between the ages of 4 and 13 years, although it can be seen in adolescents and adults (53). Onset is often subacute and presents with some combination of headache, lethargy, behavioral
and cognitive disturbances, personality changes, progressive clumsiness, and ataxia. Most cases exhibit hemiparesis or asymmetric double hemiparesis, variously combined with aphasia, visual disturbances, dysarthria, oropharyngeal dysfunction, bilateral pyramidal signs, ataxia, or pseudobulbar manifestations (271,273,274,275,276). Elevated intracranial pressure with papilledema is occasionally encountered. Unlike ADEM, there is usually no history of prodromal illness or fever (53).
and cognitive disturbances, personality changes, progressive clumsiness, and ataxia. Most cases exhibit hemiparesis or asymmetric double hemiparesis, variously combined with aphasia, visual disturbances, dysarthria, oropharyngeal dysfunction, bilateral pyramidal signs, ataxia, or pseudobulbar manifestations (271,273,274,275,276). Elevated intracranial pressure with papilledema is occasionally encountered. Unlike ADEM, there is usually no history of prodromal illness or fever (53).
Recently, optic neuritis has been reported as a novel mode of presentation in Schilder disease (277)
Diagnosis
The subacute onset of focal neurologic signs and increased intracranial pressure often suggests a space-occupying lesion, namely a brain tumor or an abscess (278,279). The CSF protein levels are usually within normal range, as is the CD4/CD8 ratio. Oligoclonal bands are detected in some cases (262,276).
Extensive hypodense areas are typically seen on computed tomography (CT). MRI shows massive involvement by either single, albeit large, or multiple areas of cerebral white matter disease consistent with demyelination. Multiple lesions in Schilder disease are characteristically bilateral, but exceptionally multiple unilateral lesions may occur (278). The typical large lesions are usually clearly visible as areas of hyperintense bright signal on T2-weighted images. Some lesions have a tumefactive (tumor-like) cystic appearance, with a peripheral rim enhancing with gadolinium (122,271,276,278,279).
The absence of significant perilesional edema, the irregular and incomplete ring enhancement, and the discrepancy between size of the lesions and the associated mass effect may help differentiate Schilder disease from a neoplastic or pyogenic process (278). As a rule, neuroimaging studies exclude the diagnosis of a tumor or abscess and point to a demyelinating process. The diagnosis of diffuse sclerosis can be made with a high degree of certainty because no other demyelinating condition can progress relatively rapidly to produce edema.
It is important to emphasize that the diagnosis of Schilder disease cannot be made unless adrenoleukodystrophy (ADL) has been ruled out by analysis of the long-chain fatty acids of plasma cholesterol esters (262,280) (see Chapter 3). ADL, which occasionally can progress relatively rapidly, can be distinguished by the presence of very long chain fatty acids in the serum. In addition, ring enhancement distinguishes the lesions of Schilder disease from the myelin abnormalities observed in ADL, which is usually found in the parieto-occipital white matter (281).
MDS must be included in the differential diagnosis in young patients with a brain tumor with atypical neuroimaging features (279,282). Many of these lesions are subjected to open craniotomy and resection because of the suspicion of tumor (278,279,282,283). Biopsy and frozen sections of these are often misinterpreted as astrocytoma. However, the inflammatory nature of the lesion dominated by macrophages, perivascular mononuclear cuffs, and reactive astrocytes is more easily recognized in paraffin-embedded material, especially with the aid of immunohistochemical markers (50,278).
Treatment
Immunosuppression with corticosteroids (methylprednisolone) or with a combination of cyclophosphamide and adrenocorticotrophic hormone (ACTH) has been reported to induce rapid, often dramatic and unequivocal improvement in the majority of cases, with complete or near-complete resolution of the MRI lesions (275,276). In the series by Barth and coworkers, all 5 patients did well after corticosteroid therapy (273).
The rapid early improvement of the clinical manifestations and neuroimaging changes after administration of corticosteroids is a characteristic feature of Schilder disease. Maximal recovery may require weeks or months of treatment and can often be incomplete. The time for discontinuing steroids is not clear-cut and may be dictated by the clinical picture. It may vary from 4 to 6 months (279). The benefit of adding intravenous immunoglobulin to the regimen in patients who attained only partial response to corticosteroid therapy is uncertain (279), and more studies are needed in this regard. The precise effect of corticosteroids in achieving maximal recovery or preventing progression to MS is unclear. Although some uncertainty exists about the optimal dose, it is generally agreed that high doses should be administered intravenously for 3 to 5 days followed by an oral taper. Although the MRI appearances usually improve, abnormalities may persist for years, especially if there is necrosis or cavitation in large tumor-like lesions. In some cases surgical decompressive aspiration of large lesions may improve the recovery (284).
The majority of patients appear to experience prolonged remissions after treatment; however, occasionally there are recurrences (275). As in MS, it is important to distinguish exacerbation caused by tapering of steroids from true recurrences.
Garell and coworkers suggested that there may be two clinical subsets of the disease: (a) a self-limiting, monophasic type, in which patients do well after the first brief episode, and (b) a progressive type, which is less responsive to treatment and may result in severe neurologic deficits or death (279). It is unclear whether these apparently divergent courses in the natural history of the disease represent inherent variations in the nosologic spectrum of MDS or are the result of early or efficacious treatment. Interestingly, in Garell’s small series, consisting of only 3 patients, the patient who underwent gross full resection did considerably better than the 2 patients who were treated only medically (279).
Neuromyelitis Optica (Devic Disease)
In 1894, Eugene Devic described the case of a 45-year-old woman suffering from bilateral optic neuritis associated with acute transverse myelitis (285). Later, his student F. Gault reviewed 16 patients described in the literature with the same symptoms (285a). Since then, the association of optic neuritis and acute transverse myelitis is known as the syndrome of neuromyelitis optica (NMO) or Devic disease, where both clinical manifestations appear simultaneously or within several weeks or months of one another (53,64,286,287).
NMO is a rare condition in the Western world, although it is not uncommon in Asia (50). It is more frequent in adults (median age of onset is in the fourth decade) and more prevalent in women (288), but it can present in children as well (289,290,291,292). Whether NMO is a subtype of MS or a distinct entity is controversial (292). For some authors, NMO is a form of MS, probably modified by histocompatibility antigens or by external factors (53). But it can also be associated with ADEM, systemic lupus erythematosus (SLE), and Sjögren syndrome, infectious diseases, and immunizations (286,288,289). Although familial cases have been reported, NMO is generally a sporadic disease (288).
Pathogenesis
The immunopathogenesis of NMO is unclear. Inflammation plays a major role in NMO, where BBB damage and inflammatory cells prevail, although these changes can fluctuate. The lesions are mostly inflammatory at onset and may undergo necrotizing changes over time (293). Stansbury proposed in 1949 that perivascular inflammation is the initial stage in the pathogenesis of NMO lesions (294). Autopsy studies of patients with NMO have shown abnormal vasculature in the spinal cord (295). The latter may be a target for autoimmune inflammation, with participation of immunoglobulins, activated complement (C9 neoantigen), and macrophages immunoreactive for myelin proteins, including MOG; in addition, T cells and monocytes/macrophages may contribute to the myelin damage (142,286,288). Moreover, eosinophils may play a significant role in the destructive inflammatory process of NMO (142).
Pathology
The most typical neuropathologic features of the lesions are demyelination associated with inflammatory necrosis, a topographic distribution restricted to the anterior part of the optic tract (optic nerve and usually the chiasm) and the spinal cord without any signs of other lesions elsewhere. The spinal lesions are located in one or several segments. They sometimes extend to the whole spinal cord, but usually are not multicentric (296). The lesions progress through a series of stages (294):
Acute inflammation with prominent perivascular exudates. Subsequently, inflammatory infiltrates consist mainly of macrophages and microglia, with B lymphocytes also prominent, but few CD3+ or CD8+ T lymphocytes. Early lesions are also characterized by the combined presence of eosinophilic and granulocyte perivascular infiltration (142).
Tissue destruction and demyelination.
Coalescence of small lesions into larger ones, involving gray and white matter, and necrosis.
Reactive microglial changes.
Astrocytosis and formation of glial scars. Glial scarring is less frequent and usually only partial, in contrast to typical MS plaques.
The presence of hyalinized medium-sized spinal cord arteries is very characteristic (288,291). The selective localization of NMO lesions may be explained by the vulnerability of the spinal cord and optic nerve to antibody-mediated injury due to inherent susceptibility of the BBB at these sites or less likely by the fact that the latter regions of the neuraxis may harbor anatomically restricted neural or vascular antigen(s) (142).
Clinical Manifestations
Clinically, ON attacks are generally severe and exhibit poor recovery; a minority of patients experience simultaneous bilateral ON. Myelitis attacks are often fulminant, bilateral, complete acute transverse myelitis (ATM) and accompanied by pain and a great degree of residual neurologic impairment (287,288,296). The clinical course is monophasic in about 35% of patients, where there is a single episode of ON and ATM and several years of follow-up that reveals no further exacerbations. Most patients experience a relapsing course, where ON and ATM may be many weeks or even years apart but attacks recur over the next months or years (288,297); 55% of relapses occur within the first year after the initial clinical event, 78% at 3 years, and 90% at 5 years (298).
Diagnosis
The diagnosis is based on the typical association of ON and ATM (see Table 8.5)
The differential diagnosis with connective tissue diseases and MS may be difficult in some cases at the time of the first clinical attack. Early diagnosis and treatment are very important to reduce morbidity of NMO (288,299). Recently, Lennon and collaborators (299) described NMO-IgG, a serum autoantibody against a putative target autoantigen of NMO, associated with both the subarachnoid glia limitans and Virchow-Robin spaces and the
extracellular matrix or parenchymal-penetrating microvessels in the CNS. This antibody was detected in almost three-fourth of patients with NMO, in nearly half of those at high risk of developing NMO, and in about one-tenth of those showing ON or ATM as the initial manifestation of MS. However, it was negative in all patients with classic MS. The authors concluded that NMO-IgG is a specific marker autoantibody of NMO, which is useful for establishing a differential diagnosis with MS and for monitoring the progression of ONM and its response to treatment (299). Patients with ONM also have a high seropositivity for multiple markers of autoimmunity such as antinuclear antibody (ANA), extractable antinuclear antigen (ENA), and thyroid antibodies (288,289).
extracellular matrix or parenchymal-penetrating microvessels in the CNS. This antibody was detected in almost three-fourth of patients with NMO, in nearly half of those at high risk of developing NMO, and in about one-tenth of those showing ON or ATM as the initial manifestation of MS. However, it was negative in all patients with classic MS. The authors concluded that NMO-IgG is a specific marker autoantibody of NMO, which is useful for establishing a differential diagnosis with MS and for monitoring the progression of ONM and its response to treatment (299). Patients with ONM also have a high seropositivity for multiple markers of autoimmunity such as antinuclear antibody (ANA), extractable antinuclear antigen (ENA), and thyroid antibodies (288,289).
TABLE 8.5 Diagnostic Criteria of Neuromyelitis Optica (Devic Disease) | ||||||||
|---|---|---|---|---|---|---|---|---|
| ||||||||
The CSF is often abnormal with mild elevation of protein and pleocytosis, including neutrophils, up to 3,000 cells/mm3. Pleocytosis can vary depending on the phase of the disease, being relevant during the acute symptomatic event(s) and normalizing during the stationary phase. Oligoclonal bands may be present, but less often than in typical MS, and, if they are present, they usually disappear in the course of the disease, contrary to what happens in MS (286,289,290,293). In patients with relapsing NMO total IgG concentration is elevated, as in MS patients, but the percentage of IgG1 and IgG1 index are increased only in patients with MS; these findings suggest less Th1 immunity in relapsing NMO and may also explain the rarity of oligoclonal bands in patients with this disease (300).
Brain MRI in NMO patients shows no focal white matter lesion suggestive of MS, whereas spinal cord MRI displays extensive cervical or thoracic confluent, longitudinally extensive lesions, usually longer than two vertebral segments. They are T1 hypointense, which distinguishes them from those described in MS, which are hyperintense in T2-weighted sequences and enhance with gadolinium (288,289,290,293).
In selected cases, leptomeningeal biopsy shows increased vascularization and thickened hyalinized vessel walls (291).
Treatment
The treatment of choice for acute attacks in NMO is intravenous methylprednisolone for 5 days followed by prednisone taper. In patients refractory to corticosteroids, intravenous IVIG has been used with some success (286,288,291). Plasmapheresis, in a randomized, double-blind, crossover trial in patients with idiopathic inflammatory demyelinating disease, showed moderate or marked improvement in 6 of 10 patients with severe, corticosteroid-refractory NMO (301).
Case series of NMO patients suggest that azathioprine and prednisone may reduce relapse frequency and protect optic nerve and spinal cord function more effectively (302). The use of other immunosuppressive drugs like cyclophosphamide and mycophenolate mofetil has been limited (286,288). The role of immunotherapy with interferons or glatiramer acetate is unclear, but anecdotal evidence has been discouraging (288). Based on knowledge about the immunopathogenesis of NMO, B-cell modulators should be investigated as possible therapeutic agents (288).
Prognosis
The prognosis of patients with NMO has been well studied in adults, and is related to the severity and clinical course of the disease (288,289,290). In the experience of the Mayo Clinic, patients with monophasic NMO have more severe acute ON and ATM, but the long-term neurologic impairment and disability is significantly less than in patients with a relapsing course. Although 22% of them may remain legally blind at least in one eye, more than 50% recover to 20/30 visual acuity or better. Neurologic impairment is mainly related to sequelae of ATM; most patients experience at least a moderate degree of both limb weakness and sphincter dysfunction. Five-year survival in this group is 90%, and the cause of death is unrelated to NMO or its complications (288). A relapsing course of NMO is more likely to occur if patients are of female gender, are older at disease onset, and have less severe motor impairment with the first ATM attack and there is a longer interval between the first and the second attacks. The prognosis is worse in these patients: at 5 years of follow-up more than 50% will be legally blind in at least one eye. In addition, with each relapse the motor disability increases. Life expectancy is seriously affected: Almost one-third of patients die secondary to recurrent myelitis with respiratory failure and attendant medical complications. The presence of autoimmune disease, the degree of motor recovery after
the initial myelitis event, and a higher attack frequency in the first 2 years of disease all predict a shorter survival (288,292,297). Other series of patients with NMO reported similar prognostic data (289,290).
the initial myelitis event, and a higher attack frequency in the first 2 years of disease all predict a shorter survival (288,292,297). Other series of patients with NMO reported similar prognostic data (289,290).
Encephalitis Periaxialis Concentrica (Baló Disease)
Encephalitis periaxialis concentrica was first described in 1927 by Josef Baló in Hungary and was published in the English-language literature in 1928 (303). The characteristic pathologic findings are alternating rings of myelin preservation or remyelination and myelin loss, consistent with demyelination, involving the cerebral hemispheres, cerebellum, brainstem, spinal cord, and optic chiasm (303,304). Lesions show sharp edges, and in almost all cases there are separate small plaques of demyelination (3). Both findings are typically seen in MS, and, therefore, Baló disease is now considered a variant of MS (53).
The exact mechanism for the peculiar configuration of the plaques is unclear, but it has been suggested that it represents a local phenomenon (3). Although the initial triggering event is unknown, a centrifugally spreading band of lymphocytes emanates from the initial site. The polarity of the demyelinating bands suggests that the demyelinating activity is periodically reactivated and then fades in strength as it migrates from the center (304). Candidate modulators of immune activity include the cytokines, some of which undergo periodic level fluctuations (304,305).
The disease appears to be more common in persons of Asian descent, suggesting a genetic predisposition (3,304). It usually has an acute or subacute onset and follows a monophasic course over a period of weeks or months, suggesting a space-occupying lesion (3,304). Frequently, the clinical picture is indistinguishable from that of ADEM (see later discussion) (53).
MRI plays a central role in antemortem diagnosis of this rare disease. The typical findings consist of concentric rings or a whorled appearance on T2-weighted and contrast-enhanced T1-weighted images, which corresponds to pathologic findings (304,306,307). Serial MRI studies demonstrated that concentric lesions do not occur simultaneously, but develop in a stepwise and distinctive centrifugal fashion (308). Brain MRS performed in some patients shows increased choline peak and decreased N-acetylaspartate peak, findings similar to the ones described in MS patients (71,197). In some unclear cases, a brain biopsy may be necesary to exclude tumefactive processes such as neoplasm or abscess (304).
High doses of intravenous corticosteroids produce significant improvement of the neurologic symptoms and signs (304,307). Recent reports show a benign course of the disease, without relapses after responding to treatment (304,309,310). This is in contrast with the classic description of patients with a progressive and fatal course diagnosed at autopsy. Therefore, it is likely that benign cases were missed or misdiagnosed before the availability of MRI (304).
Acute Disseminated Encephalomyelitis
Acute disseminated encephalomyelitis (ADEM) is an immune-mediated inflammatory disorder of the CNS, which is commonly preceded by an infection and predominantly affects the white matter of the brain and spinal cord (64,188,311,312,313,314).
Although the literature indicates that ADEM is not frequent, it is difficult to establish its real incidence because only isolated cases or small series of patients are usually reported. Leake and colleagues reported an overall incidence of 0.4 in 100,000 per year (315). The incidence of ADEM quadrupled during 1998 through 2000 as compared with earlier years, but this may be attributed to the widespread use of MRI, which has facilitated a more accurate identification of this condition, otherwise frequently diagnosed as “acute nonspecific meningoencephalitis” (188). It has been estimated that ADEM represents approximately one-third of all the patients diagnosed as having encephalitis in the United States (316,317).
ADEM can occur at any age, but it is much more frequent in children probably because of a higher exposure to infections and immunizations. In our experience, the mean age of patients is 5 years, with a male preponderance (188). The presentation may be acute or subacute, and it is typically monophasic, although recurrent or relapsing forms have been reported (190).
Historical Aspects
Illnesses recognizable as ADEM were first described in the late nineteenth century by Osler and others, who were particularly struck by the occasional child who showed remarkable recovery from severe, acute, multifocal encephalitic illnesses. Many cases occurred in association with epidemics of viral illnesses that spread through Europe in the wake of World War I. The characteristic pathology was described almost simultaneously in the late 1920s and early 1930s in children who had died of ADEM after measles, chickenpox, influenza, smallpox, and/or vaccinations (53,313,318).
Over the years, this disorder has received different names, reflecting different salient aspects of the disease:
Triggering events: postinfectious or postvaccinial encephalomyelitis (319).
Histopathologic features and distribution of lesions: acute perivascular myelinoclasia (320), perivenous encephalitis (321), disseminated vasculomyelinopathy (322).
Based on our clinicopathologic understanding of the disease, ADEM is probably the most appropriate nosologic designation because the precipitating infective event may be absent and the various underlying disease mechanisms are unclear.
Pathogenesis
The pathogenesis of ADEM is thought to be due to disseminated multifocal inflammation and patchy demyelination associated with autoimmune mechanisms in the CNS (53,64,326). Attempts to recover viruses or to demonstrate the presence of viral particles or antigens in the lesions have been unsuccessful. The absence of the typical pathologic findings seen in viral infections indicates that a direct viral invasion of the CNS is not the cause of the disease. The presence of a “silent” (clinically asymptomatic) interval between an antecedent of infection or immunization and the beginning of the encephalopathy, along with the presence of pathologic changes similar to the ones observed in EAE, supports an autoimmune mechanism (2) (see later discussion).
The occurrence of ADEM in humans exposed to rabies vaccine contaminated with brain tissue lends credence to the usefulness of EAE as an experimental model for ADEM and MS (2,312,313,327). The autoimmune hypothesis indicates that T cells directed against viral or bacterial antigens recognize sequences of amino acids shared with myelin proteins (2,328). Activated T cells cross the BBB, facilitating the recruitment and migration of other inflammatory cells, which would contribute to the demyelinating process. Target antigens include basic myelin protein (MBP), proteolipid protein (PLP), myelin oligodendrocyte protein (MOP), myelin associated glycoprotein (MAG), oligodendrocyte basic protein, and others (2,64,311,312,329). Humoral immunity may also play a role. Dale and colleagues (330) demonstrated autoantibodies reactive against putative basal ganglionic antigens in poststreptococcal ADEM associated with dystonia and lesions in the basal ganglia. Autoimmunity can be triggered by several mechanisms, including molecular mimicry, bystander activation, epitope spreading, and “mistaken self” (331). The degree to which these different autoimmune mechanisms are operative in ADEM is not known (313).
The exact molecular mechanisms that cause death of oligodendrocytes in ADEM and its variants are not known; however cytokines, chemokines, and adhesion molecules may collectively contribute to the pathogenesis of inflammatory encephalomyelitis (311,312). TNF-α is considered an important factor in the pathogenesis of EAE (332). It has been suggested that upregulation of Fas ligand (FasL) on autoreactive infiltrating T cells together with upregulation of Fas in resident oligodendroglial cells may account for neural tissue damage via an apoptotic pathway (333).
Genetic susceptibility explains why encephalomyelitic complications develop only in a small percentage of patients who have infections or receive immunizations Among candidate polymorphic MHC and non-MHC genes that contribute to disease susceptibility, including those that encode for effector (cytokines and chemokines) or receptor molecules within the immune system, human leukocyte antigen class II genes have the most significant influence (312,334). Active nitrogen species are overproduced in EAE and nitric oxide has been shown to mediate the death of oligodendrocytes (335). Other suggested mechanisms include oxidative stress–induced cell death of premature oligodendrocytes (336) and excitotoxicity (337).
Pathology
Macroscopic examination of the brain can be essentially unremarkable, although frequently there is edema, with signs of cerebral congestion. The distinguishing histopathologic feature of ADEM is demyelination with perivascular, particularly perivenous inflammation, involving predominantly white matter areas of the cerebral hemispheres, brainstem, cerebellum, spinal cord, and optic nerves (312). The inflammatory process is characterized by a perivascular infiltration of inflammatory mononuclear cells (lymphocytes and monocytes), typically around veins and venules and reactive microglial proliferation. Not infrequently, instances of frank vasculitis, with or without segmental necrotizing changes, marked by transmural inflammatory involvement are encountered. There is associated vasogenic edema, which causes variable degrees of brain and spinal cord swelling. It is significant that the inflammatory process involves both white and gray matter, although the former exhibits more prominent involvement and more severe changes. Within the lesions there is myelin fragmentation with relative preservation of axons, but there may be substantial axonal damage (338). The lesions can be confluent, forming large demyelinating areas. The leptomeninges and the Virchow-Robin spaces show cellular infiltrates of lymphocytes, plasma cells, and, occasionally, neutrophils during the initial stages of the disease. In the late stages, the inflammatory response is replaced by fibrillary gliosis (339).
Clinical Manifestations
Neurologic manifestations begin 3 days to 4 weeks (mean 12 days) following a precipitating event, which can be identified in as many as three-fourths of patients in the experience of one of us (S.N.T.) (Table 8.2) (183).
According to the literature, the viral agents most frequently related to ADEM are influenza A and B, parainfluenza, mumps, rubella, varicella, herpes simplex type 1 (HSV), human herpesvirus-6, Epstein-Barr virus, cytomegalovirus (CMV), hepatitis A and B, and coxsackie virus (311,312,313,315,328,340,341,342,343). Measles infection poses the highest risk for development of ADEM: 1 in 400 to 1 in 1,000 cases, as compared with 1 in 10,000 cases for varicella and 1 in 20,000 cases for rubella (342,344). Less frequently, bacterial infections are implicated in the development of ADEM, including Mycoplasma pneumoniae, Borrelia bugdorferi, Chlamydia, Campylobacter, Rickettsia rickettsii, Streptococcus pyogenes, Streptococcus β-hemolytic group A, and Bartonella henselae (342,345,346,347). Immunizations linked to the development of ADEM are those against measles, mumps, rubella, diphtheria-pertussis-tetanus (DPT), varicella, mumps parotitis, rubeola, influenza, Japanese encephalitis type B, and poliomyelitis (311,312,313,346,348,349,350,351,352). However, the only pathologically proven causal association has been with the old antirabies vaccine, which it is not currently used (327).
According to the literature, the viral agents most frequently related to ADEM are influenza A and B, parainfluenza, mumps, rubella, varicella, herpes simplex type 1 (HSV), human herpesvirus-6, Epstein-Barr virus, cytomegalovirus (CMV), hepatitis A and B, and coxsackie virus (311,312,313,315,328,340,341,342,343). Measles infection poses the highest risk for development of ADEM: 1 in 400 to 1 in 1,000 cases, as compared with 1 in 10,000 cases for varicella and 1 in 20,000 cases for rubella (342,344). Less frequently, bacterial infections are implicated in the development of ADEM, including Mycoplasma pneumoniae, Borrelia bugdorferi, Chlamydia, Campylobacter, Rickettsia rickettsii, Streptococcus pyogenes, Streptococcus β-hemolytic group A, and Bartonella henselae (342,345,346,347). Immunizations linked to the development of ADEM are those against measles, mumps, rubella, diphtheria-pertussis-tetanus (DPT), varicella, mumps parotitis, rubeola, influenza, Japanese encephalitis type B, and poliomyelitis (311,312,313,346,348,349,350,351,352). However, the only pathologically proven causal association has been with the old antirabies vaccine, which it is not currently used (327).
Initially the patient may complain of nonspecific symptoms such as headache, low-grade fever, myalgia, and malaise. These are followed by a rapid onset of overt neurologic manifestations including acute encephalopathy coupled with a triad of focal neurologic deficits (hemiparesis, quadriparesis), ataxia, and change in mental status (sleepiness, stupor, or coma). Other symptoms and signs include cranial nerve involvement, meningismus, convulsions, migraine, myelopathy, optic neuritis, aphasia, involuntary movements, and paresthesias (64,188,311,312,313,353,354).
Acute severe combined demyelination is characterized by combined findings of central and peripheral demyelination. These patients fulfill the clinical and electrophysiologic diagnostic criteria of Guillain-Barré syndrome, they develop acute change in mental status and multiple cranial nerve palsies, and their neuroimaging studies show white matter abnormalites characteristic of ADEM (355,356).
Diagnosis
The diagnosis of ADEM is based on MRI evidence of multifocal white matter demyelination of a patient in whom there is acute onset of neurologic dysfunction after a latent period preceded by a systemic infection, usually viral, or an immunization (64,188,311,312,314,315,326,357,358).
Immunologic Studies
The peripheral blood counts may be within normal range, although they can also show leukocytosis with lymphocytosis. The CSF is essentially unremarkable in one-half of the patients, whereas in the other half it may show a mild to moderate pleocytosis, rarely, though, above 100 cells/mm3. Glucose is usually within normal range and protein is moderately elevated (64,188,311,312,314,315, 326,357,358).
The presence of IgG oligoclonal bands or increased IgG index in the CSF, indicating intrathecal synthesis of immunoglobulins, is a rare occurrence (311,312,313,357). In a series of 84 patients previously reported by one of us (S.N.T.), only 2 patients showed presence of oligoclonal bands; both patients had developed ADEM following HSV encephalitis (188). The finding of increased CSF MBP is an indicator of the destruction of myelin and depends on the timing of performing the spinal tap and on the extent of the demyelinating lesions and has no clinical diagnostic value. High levels of IL-6 (311) and low levels of IL-10 and TNF-α (315) have recently been reported in the CSF of ADEM patients.
A recent study by Yoshikawa and colleagues (359) found low levels of hypocretin in the CSF of ADEM patients that correlated with excessive daytime sleepiness during the course of the disease. The authors suggested that a dysfunction of hypothalamic hypocretin-peptide neurotransmission is involved in the altered state of alertness in these patients.
Neurophysiology
Neurophysiologic studies show nonspecific abnormalities. The EEG performed during the acute phase of the disease shows generalized or focal slowing, with high-amplitude theta and delta waves (188,311,312,326). The observed prolonged latencies in the evoked potentials are nondiagnostic because they can be seen in other acute encephalopathies. However, they correlate well with the clinical and neuroimaging data and can be useful in evaluating the course of the disease. It is important to emphasize that the evoked potentials do not reveal subclinical involvement of functional systems in ADEM, in contrast to MS (311,312).
Anatomic Neuroimaging
MRI examination of the CNS is the investigation of choice for establishing the diagnosis of ADEM (64,315, 360,361), which should be suspected on the basis of the clinical history and neurologic evaluation. Demyelinating lesions are hypointense on T1 and inversion-recovery sequences and hyperintense on T2, proton density, and FLAIR images. Lesions are usually asymmetric and variable in number and size (187,213,311,312,313,326, 357,362,363). The absence of periventricular changes is one of the key features that help to distinguish ADEM from a first clinical presentation of MS (313,353). The lesions involve the deep white matter with variable compromise of the subcortical white matter of the cerebral hemispheres, cerebellum, brainstem, and spinal cord. However, cortical and deep gray matter involvement is a commonly reported finding (189,313,364). The corpus callosum is usually not
involved in ADEM; infrequently, its involvement has been reported, suggesting extensive lesion load (312,353). Lesions may not enhance after the administration of gadolinium, or, more often, may enhance in a homogeneous fashion, which indicates that they are in the same phase of progression (365,366). Different patterns of enhancement can be observed, including ring shaped, nodular, incomplete ringlike, and complete-ring shaped (367). Although multifocal or disseminated lesions are characteristic of ADEM, there have been instances with solitary CNS lesions (122,368,369).
involved in ADEM; infrequently, its involvement has been reported, suggesting extensive lesion load (312,353). Lesions may not enhance after the administration of gadolinium, or, more often, may enhance in a homogeneous fashion, which indicates that they are in the same phase of progression (365,366). Different patterns of enhancement can be observed, including ring shaped, nodular, incomplete ringlike, and complete-ring shaped (367). Although multifocal or disseminated lesions are characteristic of ADEM, there have been instances with solitary CNS lesions (122,368,369).
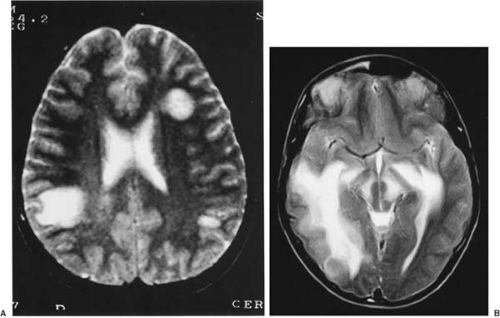 FIGURE 8.4. A: Acute disseminated encephalomyelitis (ADEM). Axial T2-weighted magnetic resonance imaging (MRI) showing small, bilateral, hyperintense lesions in the periventricular white matter. B: ADEM. Axial T2-weighted MRI with extensive bihemispheric demyelinating lesions involving the white matter and basal ganglia, extending into the mesencephalon. |
Tenembaum and colleagues (188) developed a classification of MRI lesions in ADEM, which include the following groups: A (62%), with small demyelinating lesions (less than 5 mm) (Fig. 8.4A); B (24%) with larger demyelinating plaques (greater than 5 mm or confluent), an asymmetric distribution, and variable tumefactive effect (“pseudo-tumoral ADEM”) (Fig. 8.4B); C (12%), with additional, symmetric, bilateral thalamic involvement; and D (2%): acute hemorrhagic encephalomyelitis (HAEM) or Hurst disease, with large plaques showing some evidence of hemorrhage.
The diffusion-weighted imaging (DWI)-MRI technique adds to the diagnostic power of MRI in patients with ADEM, and may help to elucidate different overlapping phases in CNS inflammation (362,370,371,372). Diffusion tensor MRI (DT-MRI) measures diffusibility or microscopic random translational motion of molecules and water, which provides information about the orientation, size, shape, and geometry of brain structures. DT-MRI of the basal ganglia has demonstrated that patients with ADEM have a high mean diffusibility (microscopic random translational motion of molecules and water), in contrast to MS patients, suggesting that deep gray matter tissue damage occurs in ADEM patients. DT-MRI may be useful for establishing a differential diagnosis between ADEM and MS (373).
Functional Neuroimaging
Studies with quantitative proton MR spectroscopy reveal low levels of N-acetylaspartate (NAA), without increase in choline, and elevated lactate within the regions of prolonged T2 MRI signal, which recover with normalization of clinical and MRI findings (369,371,374).
99m Tc-HmPAO SPECT studies have shown areas of hypoperfusion that are more extensive than the MRI lesions (375,376,377,378). SPECT also reflects better the clinical course than the time course of MRI abnormalities; in spite of
MRI becoming normal, SPECT detects persistent cerebral circulatory impairment probably contributing to cognitive and language deficits observed in these patients (376). Similarly, PET scan demonstrated global decreased cerebral metabolism in both hemispheric white and gray matter in a case where MRI showed a focal right frontoparietal demyelinating lesion (378).
MRI becoming normal, SPECT detects persistent cerebral circulatory impairment probably contributing to cognitive and language deficits observed in these patients (376). Similarly, PET scan demonstrated global decreased cerebral metabolism in both hemispheric white and gray matter in a case where MRI showed a focal right frontoparietal demyelinating lesion (378).
Differential Diagnosis
The differential diagnosis of ADEM is broad. In patients presenting with acute changes in mental status, motor focal findings, fever, and partial seizures one is required to rule out acute viral meningoencephalitis, in particular due to HSV. It is recommended to initiate specific antiviral treatment (acyclovir) until MRI and virologic studies confirm or rule out HSV infection (53).
The cases of ADEM with large MRI lesions with variable mass effect need to be differentiated from primary cerebral tumors or Schilder disease (262,368). MRI resolution with corticosteroid treatment in patients with a monophasic disease will support the diagnosis of ADEM but this is not entirely clear-cut given the responsiveness of cases of Schilder disease to steroid treatment.
The use of MR spectroscopy can be of value in supporting the diagnosis of gliomatosis cerebri because it shows high choline/creatine and choline/NAA ratios (379). Clinical and radiologic recovery after a pulse of corticosteroids is diagnostic of demyelinating disease (ADEM or Schilder disease) as opposed to tumor.
The metabolic leukoencephalopathies, such as metachromatic leukodystrophy, Krabbe disease, and adrenoleukodystrophy, as well as mitochondrial disorders should be included in the differential diagnosis (313). However, the clinical course in these disorders is different than in ADEM.
Isolated angiitis of the CNS and macrophage activation syndromes (i.e., familial hemophagocytic lymphohistiocytosis) should also be considered in the differential diagnosis (380).
In patients with bithalamic MRI involvement one has to rule out acute necrotizing encephalopathy of childhood, which presents with a severe, acute neurologic dysfunction and bilateral thalamic necrosis with cavitation (381,382). Patients with ADEM show complete resolution of the bithalamic lesions without cerebral atrophy or gliosis (188,383,384,385). A similar neuroradiologic pattern can be observed in patients with deep cerebral venous thrombosis (386), but a careful observation of T1 and T2 MRI images in sagittal views makes it possible to rule out this condition when not showing signal changes in the deep cerebral veins and straight sinus. Infantile striatal necrosis and other forms of encephalitis involving the basal ganglia should also be considered in the differential diagnosis of patients with bilateral bright MRI signal in the caudate and lenticular nucleus (387).
Treatment
There are no controlled clinical trials investigating therapies for ADEM. Several studies have reported spontaneous and rapid improvement in untreated patients with ADEM (342,357,364,374). Low-dose corticosteroids have no beneficial effect and may be contraindicated (313). However, the use of high doses of corticosteroids during the acute period of the disease is considered the specific treatment directed against the inflammatory immune process. The efficacy of this regimen has been shown in several reports in that it shortens the duration and severity of the encephalopathy (64,188,311,326,357,388,389). The use of intravenous pulses of methylprednisolone is particularly indicated in the clinical forms with severe impairment of consciousness or involvement of the optic nerves or spinal cord and in the neuroradiologic forms with pseudotumor-like or expansive effect (93). The recommended dose of methylprednisolone is 30 mg/kg per day for children less than 30 kg and 1 g/day for those above this weight, for 3 to 5 consecutive days. In patients with a less severe clinical picture one can use oral corticosteroids such as methylprednisone at 2 mg/kg per day or deflazacort at 3 mg/kg per day, for 10 to 15 consecutive days. In every case the discontinuation of the corticosteroids should be gradual over a period of 4 to 6 weeks.
The use of intravenous IgG has been reported in a few case studies and in a small series of children with satisfactory results, particularly in severe cases who did not respond to corticosteroids (326,358,390,391,392). The experience using plasmapheresis in the treatment of ADEM is limited (226,326,393,394,395). In our hands, plasmapheresis has been demonstrated to be a good treatment option for patients with a fulminant form of the disease who do not improve on steroids and immunoglobulin (326).
Other alternative therapies deserve to be mentioned. One patient with a fulminant form of ADEM responded to treatment with hypothermia (396). Experimental studies suggest that cyclosporine and NOX-100, a novel nitric oxide scavenger, may have a potential therapeutic application (397,398).
Medical support is an important aspect of the treatment of patients with ADEM. It should include mechanical ventilation in the cases with severe brainstem dysfunction, correction of electrolyte derangements, treatment of the fever, use of antiepileptic drugs in the cases that present seizures, treatment of neurogenic bladder if present, use of antibiotics to treat secondary respiratory,
and urinary tract infections and administration of intravenous acyclovir at 30 mg/kg per day when viral herpes encephalitis has not been ruled out (311).
and urinary tract infections and administration of intravenous acyclovir at 30 mg/kg per day when viral herpes encephalitis has not been ruled out (311).
Prognosis
In spite of the dramatic clinical and neuroradiologic presentation, ADEM is considered a relatively benign condition; a full recovery is attained by up to 90% of patients (64,188,314,326,357,358,361,399). However, high mortality rates (10% to 30%) have been reported during the first week of the disease, particularly in cases of ADEM following measles (64,313,400) and in the hemorrhagic form (188,401). The early and aggressive use of corticosteroids and improvement in the care of neurologically critically ill patients have improved the prognosis in ADEM. Patients with poor response to the treatment probably have sustained irreversible structural damage prior to the initiation of therapy.
ADEM is, by definition, a monophasic disease in approximately 90% of patients, who will never have a similar neurologic episode. However, there have been instances of patients diagnosed with ADEM who developed clinical relapses (188,311,313,326,344,380,385,399,402,403,404). A relapse is characterized by a neurologic dysfunction, usually different than the one present during the previous episode, following an interval of at least 1 month, in a patient with partial or complete recovery. The MRI shows demyelinating lesions in a different location than the one observed during the initial episode (161,313,405). However, cases of recurrence at the previously affected brain site have been reported (406). The frequency of the biphasic (a single relapse during the follow-up) or multiphasic (more than one relapse) ADEM variants ranges between 10% and 20%; recurrences usually occur within 6 months following the initial episode (188,189,399). Biphasic or multiphasic forms of ADEM need to be differentiated from steroid dependency, which is present in patients whose lesions reactivate and whose clinical symptoms recur when stopping the treatment with corticosteroids (188,402).
The clinical variants of ADEM with one or multiple relapses raises a diagnostic problem because a demyelinating disease characterized by a clinical course of relapses and remissions is, by definition, MS. It is useful to consider the following characteristics of ADEM relapses when establishing a differential diagnosis with MS (118,188,189,190): (a) The clinical relapses are polysymptomatic, (b) the clinical follow-up confirms the absence of multiple new relapses, (c) repeated MRIs do not show new demyelinating lesions and the previous plaques resolve partially or totally, and (d) the CSF does not demonstrate the presence of oligoclonal bands (326).
The possibility of long-term progression of ADEM to MS in children seems low, although there are no series with prolonged follow-up. Tenembaum and colleagues (188) followed 8 children with biphasic ADEM for 3 to 16 years and none of them had further recurrences. The series of Anlar and collaborators (399) consisted of 46 patients followed for 3 months to 10 years; 9 of them had one relapse and 4 had multiple relapses, but no patient was diagnosed with MS. In the series of Hynson and coworkers (357), 4 of 31 children had relapses and 3 showed involvement of the corpus callosum, a feature suggestive of MS, but none of them were diagnosed as having MS. Leake and colleagues (315) followed 42 children diagnosed with ADEM over a 10-year period and found that 4 of them (9.5%) were subsequently diagnosed with MS after multiple episodes of demyelination. This is in contrast to what has been reported in adults. Schwarz and collaborators (407) followed 40 patients aged 15 to 68 years (mean 35.5 years) with ADEM, of whom 14 (35%) developed clinically definite MS. In all of the patients diagnosed with MS, the second episode occurred within the first year of initial presentation. In the longest follow-up (8 years) of 11 patients with final diagnosis of ADEM, none experienced a new clinical attack, and new white matter MRI lesions were seen in only 1 patient.
Although the neurologic recovery in ADEM is good in the majority of patients and the risk of MS is low, neurocognitive deficits are common long-term sequelae. Hahn and coworkers (408) studied 6 children followed for a mean of 4 years after the diagnosis of ADEM and found impairment of tests of attention and executive functions and lower performance than verbal IQ. Similarly, Jacobs and colleagues (409) reported that children with ADEM, particularly those who sustain the illness before 5 years of age, are vulnerable to impairments in both cognitive and social domains and present a high incidence of severe behavioral and emotional problems (409).
Acute Hemorrhagic Leukoencephalitis (Hurst Disease)
This condition is a fulminant variant of ADEM. It is rapidly progressive, causing death usually in the first week of disease due to severe brain edema. However, some patients may survive with an early and aggressive treatment with corticosteroids, cyclophosphamide, and plasmapheresis (188,410,411,412)
The gross brain may be variably swollen. Histologically, there are numerous, multifocal small hemorrhages in the white matter, with sparing of the cerebral cortex and basal ganglia. However, these microhemorrhages may also become confluent and form lobar-like hemorrhages, which are not usually massive. The microscopic examination demonstrates abnormalites predominantly in the small veins and small arteries. There is necrosis of the walls of the vessels, with fibrinous exudates, perivascular edema, hemorrhage, and scanty neutrophil infiltration (119). The
demyelination is perivascular and can even be observed surrounding ostensibly normal vessels. There is substantial axonal damage (338). Genetic susceptibility is a possible determining factor (312,354).
demyelination is perivascular and can even be observed surrounding ostensibly normal vessels. There is substantial axonal damage (338). Genetic susceptibility is a possible determining factor (312,354).
Optic Neuritis
Optic neuritis (ON) is an acute inflammatory condition affecting the optic nerve (413). In the majority of cases it probably has an autoimmune pathogenesis (313). The disease may occur in isolation or in association with other inflammatory demyelinating disorders such as MS with a predilection for spinal cord involvement akin to transverse myelitis (Devic type), classical MS, ADEM, and collagen vascular diseases (53,313). Such an association is more frequently seen in older children and adolescents (414). ON has an incidence of 1 to 5 in 100,000 per year; the incidence is highest in whites, in countries at high altitude, and in spring. Individuals aged 20 to 49 years are most at risk (415). Overall, ON is rare in children, although childhood cases have been described since the late nineteenth century. In the pediatric age group the mean age of onset is 9 to 10 years, and most patients are older than 5 years, but cases as young as 21 months have been reported (416). No gender predilection is seen in prepubertal children, but there is a 2:1 female predominance after the onset of puberty (413,416,417,418).
Clinical Manifestations
About two-thirds of patients, particularly those younger than 14 years, have a history of an infectious illness within 2 weeks prior to the onset of symptoms (417,419,420). Infections linked to ON include those related to adenovirus, measles, mumps, chickenpox, EBV (53,413), human herpes virus 7 (421), human parvovirus B19 (422), Mycoplasma (423), pertussis, and Lyme disease (413). ON may also occur following immunizations, especially for measles, mumps, rubella, hepatitis B, tetanus, rabies, diphtheria, and smallpox (413).
Prodromal symptoms and signs at onset may include fever, frontal or retro-orbital headache accompanied by scintillating scotomata, and painful eye movements (53,64,419,420). In 10% of patients no pain is reported, and in the rest its severity varies, although it does not interfere with sleep (416). A sudden impairment of visual acuity typically follows (413,416,417,418,420). Initially, the disturbance may be limited to mere visual blurring with progression over several days to partial or complete visual loss (53,64,418). In some instances, the disease becomes biocular a few days or weeks after involvement of one eye. Bilateral ocular involvement is commonly encountered in younger children, as compared to mono-ocular disease, which is seen in adolescents and adults (53,413,414,416,420). In part this tendency may reflect the ability of small children to ignore unilateral visual loss (53). Light flashes (phosphenes or photopsias) might be seen by the patient on eye movement (416). Some patients have subclinical symptoms only manifested as Unthoff phenomenon (visual deterioration on getting warm or during exercise) (416).
Children with ON may also have neurologic deficits, including seizures or signs of cerebellar dysfunction (413,424).
The initial visual acuity ranges from 20/15 all the way to no light perception; however, profound visual loss at onset is more common in children than in adults (413,417). A vision of 20/200 or worse, including counting fingers, has been reported in 84% to 100% of cases in several series (416,417,420,425).
Children with ON usually also have severe visual field defects, including central and cecocentral scotomata, contraction, and other types of defects (arcuate, altitudinal, or vertical) (413,415,417,420).
Color vision abnormalities are common, although color vision testing is limited in very young children (415,420). There is loss of red vision (red desaturation) and loss of duration or variety of the flight of colors that occurs when the eye is closed after a period of bright illumination of the retina (53,64,415,420).
In cases with greater visual impairment, patients present an afferent pupillary defect (loss of the reflexive constriction of the contralateral pupil when the retina of the affected eye is illuminated) (53,64,415). Funduscopic examination is abnormal in 64% to 87% of cases and more commonly shows swelling (anterior neuritis or papillitis), which is usually seen in prepubertal children. Other, more striking abnormalities like optic atrophy, hemorrhages at the optic nerve margin, vascular tortuosity, or sheathing of veins can also be seen (53,64,413,417,418,419,420,424). A normal fundus can be observed when the optic nerve is involved proximal to the optic disc (retrobulbar neuritis). This finding is more common in children over 14 years of age (53,64,413,416,417).
Diagnosis
The diagnosis of ON in children is made on the basis of a combination of clinical and laboratory findings. The differential diagnosis in children includes optic neuropathy secondary to sinusitis, orbital cellulitis, optic nerve/optic nerve sheath tumors, neuroblastoma, leukemia, orbital pseudotumor, other demyelinating conditions, and malingering (53,413,416).
CSF examination is frequently unremarkable. However, it can show mild pleocytosis and elevated protein content (416,417,419,420,424). Immune-function studies (IgG index, IgG synthesis rate, oligoclonal bands) may be positive, but are of limited value in differentiating isolated ON from other demyelinating conditions (64,420,424). A major
reason to examine the CSF in these patients is to rule out other neurologic disorders that may cause visual disturbances and optic disc changes simulating idiopathic ON (420).
reason to examine the CSF in these patients is to rule out other neurologic disorders that may cause visual disturbances and optic disc changes simulating idiopathic ON (420).
Visual-evoked potentials (VEPs) are helpful in establishing the diagnosis in equivocal cases and in determining the degree of visual dysfunction (53,415,416). During the acute phase of the illness, VEPs are abnormal in almost all of the cases (absent, attenuated, or delayed P100). The abnormality may be present even in patients without evident clinical signs (418,426). After a few days, when the visual acuity improves, VEPs return to normal amplitudes in parallel with the recovering of vision, although decreased amplitude or prolonged latencies may remain in some patients for months or years, even with a good clinical recovery (53,415,426). VEP is superior to MRI for determining chronic optic nerve involvement (427). The combination of VEPs with pattern electroretinogram (PERG) can be useful in differentiating macular from optic nerve disorder: In retinal disorders, both the P50 (early) and N95 (late) components of the PERG are abnormal, whereas in optic nerve disorders, only the N95 component is abnormal (416).
Contrast-enhanced high-resolution CT with fine cuts through the orbits will show up most compressive lesions (416). However, MRI is preferable because it also shows any intrinsic optic nerve lesions, as arise in ON (416). Patients should receive triple-dose gadolinium-enhanced MRI of the brain and orbits. T2-weighted, FLAIR sequence, or fat suppression are also useful techniques (428,429,430). MRI shows signal abnormalities in 84% of the symptomatic and 20% of the asymptomatic optic nerves with ON (431). Disseminated abnormal MRI signals may be detected in the cerebral white matter of as many as 72% of patients (432). They may resolve, but in a subset of patients with monosymptomatic ON, MRI signal abnormalities may accumulate without causing any clinical manifestations of MS even when the patient is followed up for more than a decade (433). The use of serial MRIs following the mean cross-sectional area of the intraorbital portion of the optic nerve demonstrates a decline over time. This observation indicates that optic nerve atrophy occurs following ON and may continue to develop over the years (430).
Treatment
In adults, the Optic Neuritis Treatment Trial (ONTT) has provided the most comprehensive information regarding the treatment of acute ON (428). Based on data from the ONTT (434,435,436) and similar trials (437), there is no treatment for acute demyelinating ON that affects long-term visual outcome or visual prognosis compared to placebo. It has been suggested that such a lack of long-term beneficial effect might be because the corticosteroids were given too late to provide neuroprotection; treatment trials in the hyperacute phase are required to address this issue (416).
The most commonly used treatment, IV methylprednisolone (1 g/day for 3 days) followed by oral prednisone, may hasten visual recovery by 2 to 3 weeks when started within 1 to 2 weeks of symptom onset. In monosymptomatic, high-risk patients (two or more white matter lesions), IV methylprednisolone may also delay the onset of MS within the first 2 years. Oral prednisone alone may increase the risk of recurrent ON and should be avoided in patients with typical acute demyelinating ON (423). A Practice Parameter published by the American Academy of Neurology Quality Standards Subcommittee offers similar recommendations (438).
The treatment of pediatric ON with corticosteroids is controversial (416), considering the visual prognosis is good even without treatment regardless of laterality or localization (papillitis vs. retrobulbar) (413). As in adults, intravenous methylprednisolone is recommended (30 mg/kg per day for 3 to 5 days) to hasten recovery of debilitating bilateral visual loss (439), but it does not seem to have an impact on the final visual outcome (416,417). Relapse is frequent in children if the steroids are tapered too quickly, and therefore this should be done over a longer period of time than in adults (at least 4 weeks) (413).
In adult patients who have a severe form of ON unresponsive to corticosteroids, IVIG and plasmapheresis have been used. In a recent study, plasmapheresis was associated with an improvement in visual acuity in 7 of 10 patients with ON largely unresponsive to previous treatments (440).
Among patients with ON at high risk for developing MS (two or more white matter lesions 3 mm or larger in diameter, at least one lesion periventricular or ovoid), treatment with IFN-β1a (Avonex) following acute ON significantly reduced the 3-year cumulative probability of MS (441, 442).
Gene therapy techniques have been applied to experimental models of ON, like EAE. In this model reactive oxygen species contribute to optic nerve demyelination and free-radical scavengers such catalase prevent this process. Catalase gene delivery by using viral vectors could be a potential therapeutic strategy in the futures for patients with severe progressive ON (443).
Prognosis
The prognosis of childhood ON is excellent, particularly when it is bilateral, and the overwhelming majority of children recover their vision completely within weeks or months. The most common sequelae include optic nerve atrophy and impairments of color and stereoscopic vision (53,417,429,443,444). Permanent, severe visual loss is quite exceptional (53). Factors that are associated with a better prognosis in adults are (a) having a short acute lesion on triple-dose gadolinium-enhanced imaging, (b)
having higher VEP amplitudes during recovery, and (c) having a steep gradient of the initial improvement in visual acuity (429). In children, younger age, bilateral disease, and a normal MRI portend a better outcome (416).
having higher VEP amplitudes during recovery, and (c) having a steep gradient of the initial improvement in visual acuity (429). In children, younger age, bilateral disease, and a normal MRI portend a better outcome (416).
The risk of recurrent ON after 5 years of follow-up in the ONTT was 19% for the affected eye, 17% for the unaffected eye, and 30% for either eye. The risk was twice as great in patients who had developed MS and was also higher in those who were initially treated with oral prednisone (416,429).
A frequently asked question of concern with prognostic implications for patients with ON and their families pertains to the risk of developing MS at a later time. In adults the rate of progression varies among series, gender, geographic region, and duration of follow-up, ranging from 34% to 74% in New England (445) to 11% in Brazil (446). In children the figures vary from 4% to 26% (420,447,448,449). Most patients who develop MS following an initial episode of ON will have a relatively benign course for at least10 years (433). Factors that increase the risk of developing MS are bilateral sequential or recurrent ON, Uhthoff symptom, presence of oligoclonal bands in the CSF, and abnormal brain MRI (53,449,450). Conversely, presence of infection within 2 weeks before the onset of ON decreases the risk. Gender, age, funduscopic findings, visual acuity, and family history do not predict subsequent development of MS (420,449). Genetic predisposition may be an important factor in the progression of ON to MS. A recent study conducted in an Iranian cohort of ON patients showed that the HLA class II alleles DR2, A23, and B21 are strongly associated with developing MS (451).
Acute Transverse Myelitis
Acute transverse myelitis (ATM) is a condition characterized by the sudden onset of rapidly progressive weakness of the lower extremities, accompanied by loss of sensation and sphincter control, and often preceded by a respiratory infection (53). ATM has been recognized for more than 100 years. An excellent description of the clinical picture was given by Gowers in 1886 (452). Before then many diseases of the spinal cord were termed myelitis. Only subsequent to the description of MS by Cruveilhier, tabes by Duchenne, Todd, and Romberg, and syringomyelia by Gull and Hallopeau did this entity gain recognition. Its first description in this century is that by Foix and Alajouanine (453). In 1928, it was first postulated that many cases of acute myelitis were postinfectious (rather than infectious) because for many patients the “fever had fallen and the rash had begun to fade” when the myelitis symptoms began (454). It was in 1948 that the term “acute transverse myelitis” was used in reporting a case of fulminant inflammatory myelopathy complicating pneumonia (455).
Pathology and Pathogenesis
Pathologic abnormalities during the acute phase invariably include focal infiltration by monocytes and lymphocytes (CD4+/CD8+ T cells) into segments of the spinal cord and perivascular spaces accompanied by astroglial and microglial activation. During the subacute phase prominent monocyte/macrophage infiltration is observed. The presence of white matter changes, demyelination, and axonal injury is prominent in postinfectious myelitis. It is noteworthy that involvement of the gray matter is also marked in some cases (456,457). In patients with autoimmune disorders such as SLE, there are typical vasculitic lesions that produce focal areas of spinal cord ischemia without overt inflammation (457,458).
The pathogenesis of ATM is believed to be immune mediated. In 30% to 60% of idiopathic ATM cases, there is an antecedent respiratory, gastrointestinal, or systemic illness (456,457). In the majority of infections, mechanisms of autoimmunity, such as molecular mimicry and superantigen-mediated disease, account for the pathologic damage characteristic of ATM (456).
Molecular mimicry may occur in ATM. A variety of infectious agents possess antigenic determinants (i.e., proteins, glycolipids, proteoglycans) that resemble self-antigens in the spinal cord. Generation of cellular and humoral immune responses results in cross-reactive immune activation against cellular targets of the spinal cord itself (89,457,458). It is also possible that autoantibodies may initiate a direct injury to neurons. A particular pentapeptide sequence found on microbial agents is also present in the extracellular region of the glutamate receptor subunits NR2a and NR2b, present on neurons in the CNS, and can induce neuronal death (456,459). In addition, high levels of even normal circulating antibodies may cause damage in ATM (451). A patient with high levels of antibodies against hepatitis B surface antigen developed recurrent ATM; circulating immune complexes containing hepatitis B surface antigen were detected in the serum and CSF, and their disappearance after treatment correlated with functional recovery (460).
Another autoimmune mechanism may be mediated by the fulminant activation of lymphocytes by microbial superantigens. These are microbial peptides that can stimulate large numbers of lymphocytes, which trigger autoimmune disease by activating autoreactive T-cell clones (461)
Additionally, immune derangements involving ILs may participate in the inflammatory mechanism of ATM. Marked upregulation of IL-6 with increased NO production has been mechanistically linked to tissue injury in ATM (457).
Humoral derangements may also be involved in some cases of ATM (456). A group of patients with allergy to
house mites and who developed ATM had high total and specific serum IgE levels, antibody deposition within the spinal cord, perivascular lymphocyte cuffing, and infiltration of eosinophils (462). It was postulated that eosinophils recruited to the spinal cord degranulated and induced neuronal injury (457).
house mites and who developed ATM had high total and specific serum IgE levels, antibody deposition within the spinal cord, perivascular lymphocyte cuffing, and infiltration of eosinophils (462). It was postulated that eosinophils recruited to the spinal cord degranulated and induced neuronal injury (457).
Clinical Manifestations
ATM has an incidence of 1 to 4 new cases per million people per year, affecting individuals of all ages, with bimodal peaks between the ages of 10 and 19 years and 30 and 39 years (464,465,466) ATM is rare in childhood; in the first 10 years of life there are substantially fewer children affected than in the second decade. Most affected children are older than 5 years (465,467), although the condition can occur even in young infants (467,468). The male-to-female ratio in pediatric series varies form 0.5:1 to 0.9:1 (463,467,469); there is no familial predisposition (457).
Approximately two-thirds of diagnosed children have a history of a recent or a concurrent infection, which is more frequently observed around the summer months (53,467). Viruses etiologically linked to ATM include herpes simplex, herpes zoster, cytomegalovirus, Epstein-Barr, echo, coxsackie, hepatitis B, hepatitis A, influenza, measles, mumps, rubella, varicella, human T-cell lymphotropic virus-1 (HTLV-1), and HIV. Bacteria that may cause ATM are Borrelia burgdorferi, Mycoplasma pneumoniae, Yersenia enterocolitica, Chlamydia psittaci, Rochalimaea henselae, mycobacteria, and Listeria monocytogenes. Parasites can also cause ATM, including schistosoma, cysticercus, toxocara, and toxoplasma (470,471,472,473,474). ATM has also been associated with immunizations for rabies, tetanus toxoid, flu, measles, smallpox, and hepatitis B (53,456,467) and with SLE and antiphospholipid syndrome (458,472,475).
The time from infection to neurologic symptoms is usually 5 to 10 days (467,469). As a general rule, the rate of onset is proportional to the intensity of the initial discomfort (53). Before the onset of acute loss of spinal cord function, there are often nonspecific symptoms such as nausea, muscle aches, and fever. The latter may be present in up to 60% of patients (463,469,474). Occasionally, cases are preceded by relatively trivial blunt trauma to the spine (53). The neurologic picture is most frequently characterized by the presence of back pain and pain in the lower extremities, gait disturbance due to weakness, paraplegia, and paresthesias. Sphincter dysfunction is frequent, and it may be present in up to 90% of children, at times manifested as urinary retention. Other symptoms are neck stiffness in one-half of patients and respiratory insufficiency, particularly if there is cervical compromise, which can cause cardiopulmonary arrest (53). In a few cases a urinary tract infection may herald the diagnosis of ATM (467). The full-blown disease is reached 4 weeks after disease onset at the latest, and in more than 80% of cases the peak is seen within the first 3 or 10 days in hyperacute and acute cases, respectively (463,466,469,474); in subacute ATM, it may take several days or weeks for the maximal deficits to occur (53,467).
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


