INTRODUCTION
Ballism refers to abnormal large-amplitude, rapid, nonpatterned, often continuous, “flinging” movements of the proximal extremities, closely related to chorea. The first use of the term is credited to Kussmaul in 1885 (1), who felt the Greek work ballism (βαλλειν, to throw) captured the “gross resemblance of the involuntary movements to normal throwing activities.” The movements were noted to be almost exclusively unilateral. Greidenberg in 1882 identified the involvement of the contralateral subthalamic area (2), which contained a nucleus already identified in 1865 by Jules Bernard Luys. Subsequent case series supported this link, naming hemiballism as “the syndrome of the corpus Luysii” (3), although the exclusivity of this localization would later be called into question.
PHENOMENOLOGY
It may be helpful to differentiate the pathologic condition of involuntary ballism from voluntary “ballistic movement,” which is a term describing rapid (rather than smooth) voluntary movement (4,5). Voluntary ballistic movements are triggered by a burst of EMG activity in the agonist muscle, followed by a burst of activity in the antagonist muscle, then a second burst in the agonist muscle. They can involve any set of antagonistic muscles, such as the biceps/triceps or the flexor pollicis longus/extensor pollicis longus. Involuntary ballism includes large-amplitude movements, due to the involvement of proximal muscle groups, and usually multiple muscle groups in concert. The movements of ballism have little volitional control, and can sometimes be quite intense. This intensity of movement, combined with its acute onset, can cause one to conjure up the picture of a tempest, taking over one side of the body. “Flingings and hurlings of the upper extremities and kickings and poundings of the lower extremities, of the greatest abruptness and violence” (6) are exhibited. In an early description of the intensity of ballism, Martin (7) commented the “violence was such that the skin had been knocked off the knuckles and other parts of the affected hand, and also of the inner sides of both knees.” The duration of individual ballistic movements is typically brief, but the movements repeat, with variable muscle group involvement, and can be nearly continuous (Video 21.1). Ballism can occur at rest, but can also be triggered or amplified by voluntary or automatic movements. The movements can be partly and briefly suppressed and are attenuated or eliminated by sleep. Hemiballism, or unilateral involvement of the arm, leg, and often face, is by far the most common presentation, almost to the exclusion of all other forms. However, examples of monoballism are rarely reported in case series (6), as are bilateral ballism, which is sometimes termed “biballism” or “paraballism” (8,9). A review of the distribution of ballism in large case series reveals the upper extremity as being involved in all cases, leg involvement in 80%, and face involvement in 25% to 50% (10,11). The time course of ballism is usually acute or subacute. Even when due to an acute event, such as cerebral ischemia or hyperglycemia, the onset of the movement can be delayed for days or sometimes weeks.
The relationship of ballism to chorea is complex. Chorea is more likely to involve distal muscle groups, is typically of lower amplitude, less intense, more free-flowing. Chorea will have more interruptions than the characteristically continuous ballism. Chorea is more common than ballism, and may have a larger etiologic pool. Ballism is nearly exclusively unilateral where chorea can be focal, hemi-, or generalized. Clinical observations, however, suggest that ballism and chorea may coexist in the same patient or evolve into each other, particularly hemiballism and hemichorea. In case series where they have been distinctly identified, only 8% to 10% of cases had “pure” hemiballism without chorea or other dyskinetic movement (10,11). Hemiballism can evolve into hemichorea as a patient recovers, and the amplitude and velocity of the movements gradually attenuate. A long-running debate therefore exists as to whether they are two distinct conditions whose clinical expression overlaps (1) or if they should be lumped as belonging to a continuum (12). Given the strong connection, many authors favor using the term hemiballism to encompass the spectrum of hemiballism and hemichorea (13), widely hyphenated in the literature as hemichorea–hemiballism (HCHB).
PATHOPHYSIOLOGY AND LOCALIZATION
Instead of trying to distinguish chorea from ballism on the basis of phenomenology, some earlier clinicians adhered to a localization-centered approach to each disorder. Bucy (14) wrote that “the definition of hemiballismus rests primarily upon the pathology and not upon the clinical manifestations”. In fact, the localization of hemiballism to a lesion of the contralateral subthalamic nucleus (STN) was felt to be nearly absolute, based on pathologic correlation in autopsied cases. Martin (7) described a patient with hemiballism resulting from a contralateral hemorrhage of the STN area, and detailed 12 other cases of hemiballism published up to that time. Multiple lesions were typically present, but the STN was involved in 11 of the 12 cases, adding further support that hemiballism was “the syndrome of the body of Luys.” Detailed study of the effects of damage to the STN in primates (15,16) found that lesions involving at least 20% of the volume of the STN would produce hemiballism. Hemiballism would also occur if there was damage to the subthalamic fasciculus, the connection between the STN and the external segment of the globus pallidus (GPe). However, if the lesion was too large and involved the internal segment of the globus pallidus (GPi) or its outflow tracts to the thalamus (the ansa lenticularis or lenticular fasciculus), hemiballism was prevented. In animals with hemiballism from destruction of the STN, subsequent lesioning of GPi reduced or eliminated the ballism.
Despite the dogma that grew up around hemiballism, it soon became evident that involvement of the contralateral STN was neither necessary nor sufficient to cause the movement, as lesions outside the STN causing hemiballism were not uncommon. Postuma and Lang (13) reviewed recent literature with CT imaging or MRI of 120 cases, including 15 of their own cases. They reported that only 18% had a lesion in STN, 8% had a lesion near the STN, 53% had a lesion outside STN, and 20% had no identifiable lesion. Another large series of patients with HCHB after stroke found lesions in the STN or subthalamic area in only 30% of cases (17). Typical extra-STN localizations include the putamen, caudate, globus pallidus, thalamus, or even outside the basal ganglia and in the cortex. Also, cases exist where a presumably causative STN lesion appears ipsilateral to the movements (18–20). Pathophysiologic theories here include ipsilateral influence of STN motor control, perhaps through its bilateral connections with the pedunculopontine nucleus, and the presence of contralateral hemiparesis.
Using classic theories of basal ganglia connectivity (21), it was originally believed that lesions of the STN caused ballism due to reduced function of the indirect pathway, a release on the “brake” that ultimately results in hyperkinesia. This did not explain, however, why lesions in many other structures could produce ballism, or why a subsequent lesion in the GPi would ameliorate it. Rather than a complete absence of activity, or a simple reduction in firing rate, there is increasing evidence that ballism is caused by abnormal firing patterns of the basal ganglia, particularly the GPi. Hamada and DeLong (22) analyzed GPi firing patterns in monkeys after placing an STN lesion that caused hemiballism. They found not only a reduction in firing rate but also a change in the pattern of firing with an increase in long pauses between spikes (Fig. 21.1). Human microelectrode recording data during stereotactic pallidotomy surgery for hemiballism shows correlation of these long pauses in GPi activity with surface EMG recording of the ballistic movements (23). If it is the change in GPi firing pattern that contributes to ballism, such a change could be induced by involvement of a lesion in many different structures, perhaps explaining why ballism does not solely localize to the STN. Lesioning the GPi could therefore reduce or eliminate the abnormal firing pattern and reduce ballism.
ETIOLOGY
VASCULAR
The list of causes of hemiballism is extensive (Table 21.1). Vascular etiologies are perhaps the most common, with stroke making up 50% to 75% of larger case series (10,11). Chung et al. (17) reviewed 5,009 consecutive cases of stroke and found 27 patients with poststroke HCHB, for an incidence of 0.54% of all strokes. The Lausanne Stroke Registry reported 11 patients with hemiballism among 2,500 strokes, for a similar incidence of 0.44% (24). The causative strokes can be either ischemic or hemorrhagic (Fig. 21.2). Other vascular causes include arteriovenous malformations (25,26), moyamoya (27,28), cavernous angioma (29,30), or etiologies presumably related to hypoperfusion, like migraine (31) and polycythemia vera (32,33).

Figure 21.1. Representative samples of tonic activity in internal globus pallidus (GPi) neurons in a monkey before (A–C) and after (D–F) subthalamic nucleus (STN) lesions. Note the increased presence of long pauses between bursts in GPi activity after the STN lesion is placed. (From Hamada I, DeLong MR. Excitotoxic acid lesions of the primate subthalamic nucleus result in reduced pallidal neuronal activity during active holding. J Neurophysiol 1992;68(5):1859–1866, with permission.)
| Causes of Ballism |
Vascular
Stroke (ischemic or hemorrhagic)
Arteriovenous malformation
Moyamoya
Cavernous angioma
Polycythemia vera
Migraine
Metabolic
Hyperglycemia (mostly nonketotic)
Hypoglycemia
Hypocalcemia
Neoplastic
Primary CNS neoplasm
Metastatic lesion
Paraneoplastic
Postsurgical
Thalamotomy
Subthalamotomy
Deep brain stimulation of the STN
VP shunt
Infectious
Tuberculosis
Neurocysticercosis
Toxoplasmosis
Syphillis
Cryptococcal meningitis
Encephalitis
Other
Antiphospholipid antibody syndrome
Systemic lupus erythematosus
Sydenham’s chorea
Multiple sclerosis
Drug-induced (levodopa, phenytoin, gabapentin, oral contraceptive)
Neurodegenerative
METABOLIC
Hyperglycemia is a well-established cause of hemiballism. Diabetes mellitus was felt to be a frequent underlying factor in earlier reviews (1), and in the pre-CT and -MRI era, Bedwell (34) described a case of a woman with transient episodes of hemiballism, each associated with a rise in blood glucose. The description of a syndrome of HCHB associated with nonketotic hyperglycemia gained further notice in the 1980s with a case series reported by Rector and colleagues (35). Later, characteristic imaging findings of putaminal CT hyperdensity (36) and T1-weighted hyperintensity on MRI (37) were described.
The clinical course of hemiballism in these patients is somewhat different than when caused by cerebral ischemia. The onset is typically subacute, and the movements may improve with normoglycemia or may persist for months or, rarely, indefinitely. Hyperglycemia and the T1-weighted hyperintensity may precede the hemiballism by days or weeks in some cases. The threshold for hyperglycemia causing this syndrome is not known, with serum glucose values ranging from 169 to 1,264 mg/dL in one series (38). Solitary T1-weighted hyperintensity of the putamen is most common (Fig. 21.3), but the caudate and globus pallidus are also variably involved. There is variable diffusion-weighted imaging (DWI) abnormality, and gradient echo imaging can be normal or show mild hypointensity. There is no edema or mass effect present. The cause for the striatal T1-weighted hyperintensity has been a matter of some debate. Hyperglycemia was thought to be a predisposing factor for petechial hemorrhage (39,40), but the conspicuous restriction to the putaminal neuroanatomical borders without edema, the inconsistent presence of gradient echo hypointensity, and the persistence of the T1 changes over years make microhemorrhage a less likely cause. Instead, reactive astrocytosis with gemistocytes have been reported on autopsy and biopsy studies (41,42). One biopsied striatum identified vascular proliferation and arteriolar wall thickening with red cell extravasation (43), leading the authors to propose the term “diabetic striatopathy” given the similarities to diabetic retinopathy. However, some atypical features of this biopsied case included hyperintensity on DWI imaging and surrounding edema, which may limit the generalizability of their conclusions.
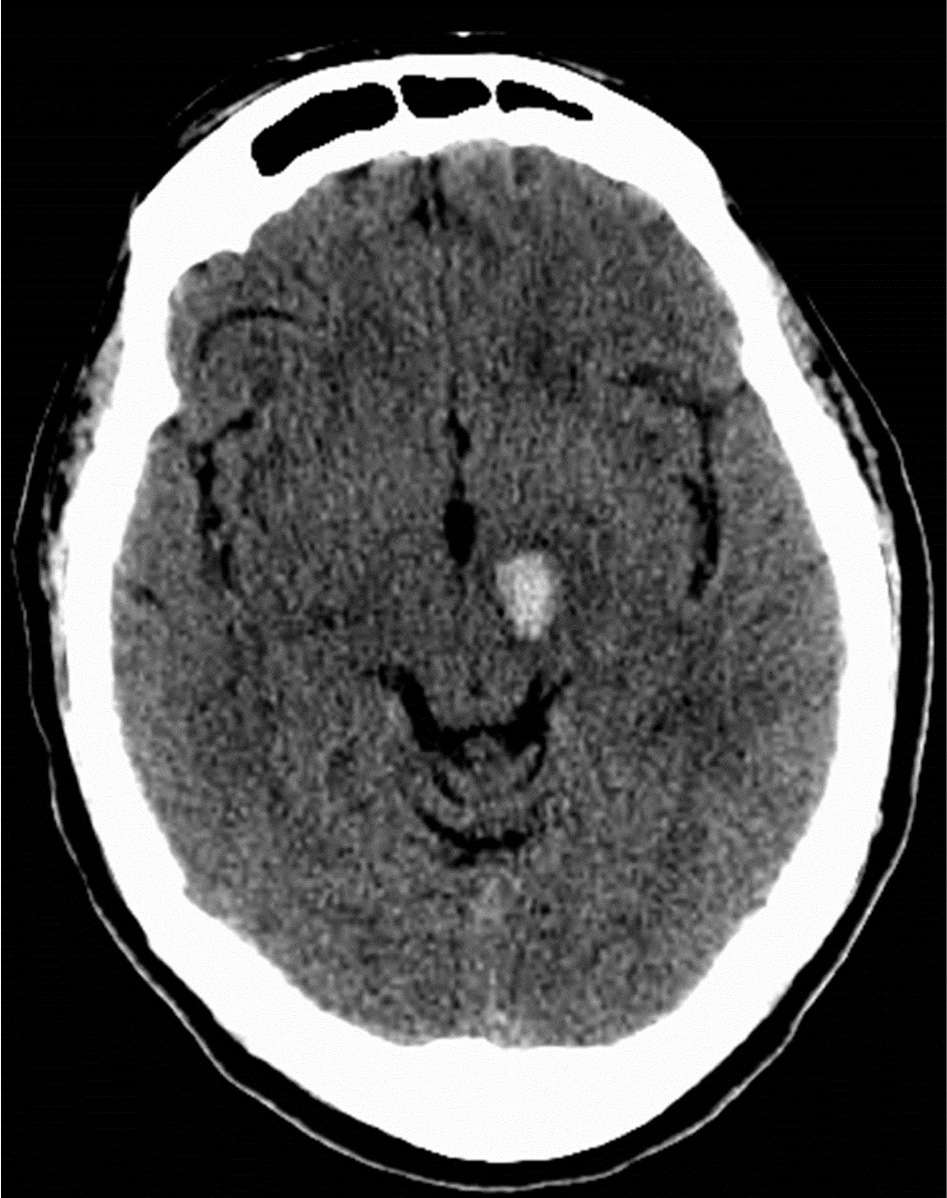
Figure 21.2. Hemorrhagic infarction involving the left subthalamic nucleus in a 60-year-old woman with acute-onset right hemiballism.
A hyperosmolar state may be an important part of the striatal pathophysiology, related to rapid shifts in plasma glucose. Supportive evidence includes several cases of central pontine myelinolysis in association with the hyperglycemic syndrome (44,45). Additional support for causal osmotic derangement may be found in a case of hemiballism with striatal T1-weighted hyperintensity in a hypoglycemic type 1 diabetic patient (46), with recurrent episodes of nocturnal hypoglycemia (1.9–2.2 mmol/L [34.2–39.6 mg/dL]). Other reports exist of hypoglycemic hemiballism (47) or biballism (48) that resolved with glucose normalization. Hemiballism caused by hypocalcemia in the setting of hypoparathyroidism has also been reported (49).
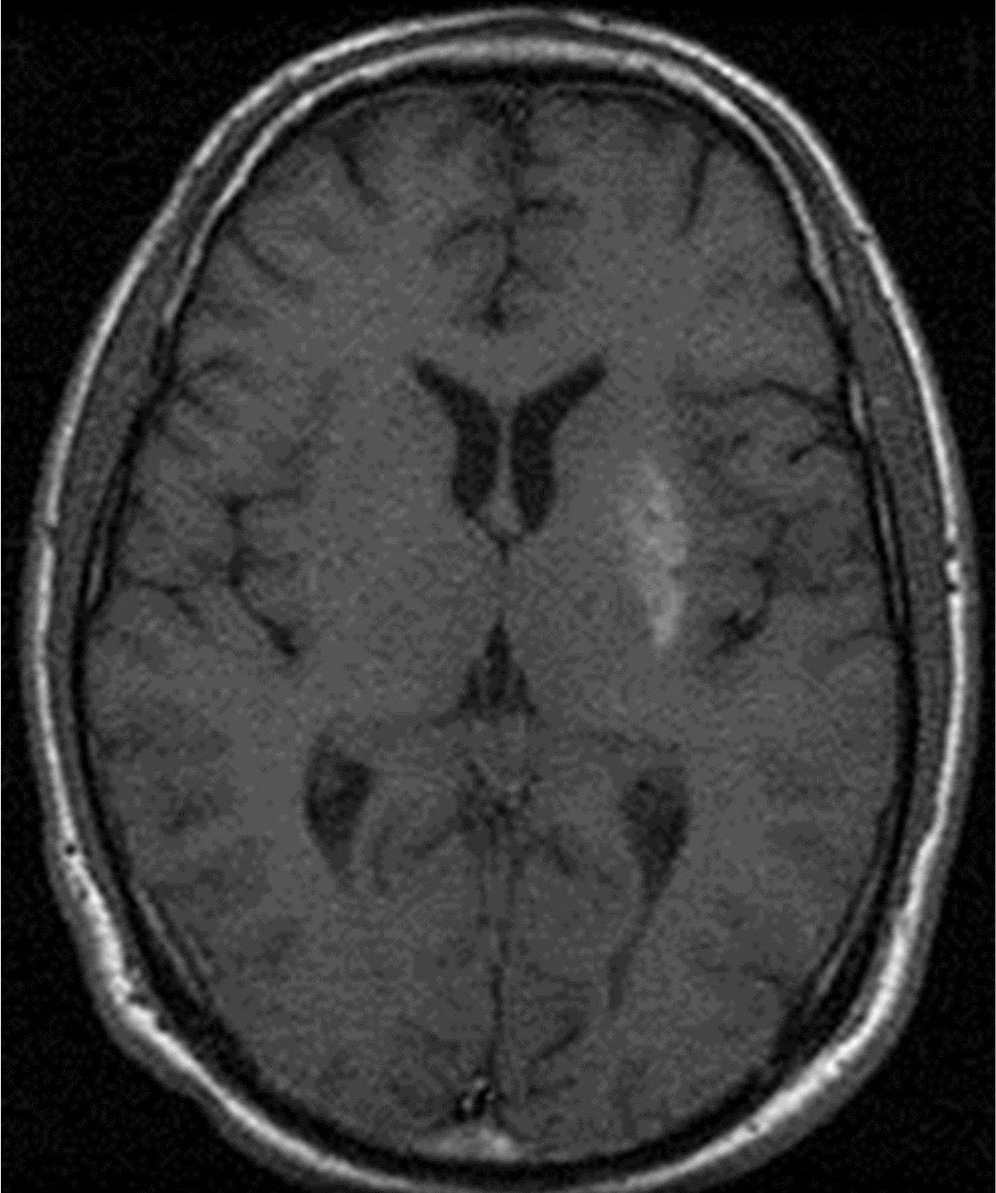
Figure 21.3. Putaminal T1-weighted hyperintensity in a 37-year-old man with hyperglycemia-induced hemiballism.




