Myoclonus is characterized by quick, involuntary muscle jerks. It is a common movement disorder, with a lifetime prevalence in 1990 of 8.6 cases per 100,000 population (1).
Phenomenologically, myoclonus may be differentiated from other movement disorders by its simplicity, its brevity, and its inability to be voluntarily suppressed. Visually, myoclonic jerks may appear identical to simple motor tics. However, tics can be voluntarily suppressed. While the tic is being suppressed, the individual often experiences an irresistible urge to move that is subsequently relieved after the tic occurs; this suppressibility and urge are absent in myoclonus. The individual movements of chorea may include myoclonic jerks. In contrast to myoclonus, however, choreiform movements continue in a constant flow, and include slower movements as well as sustained postures. Rhythmic myoclonus can appear superficially similar to tremor. Fasciculations and myokymia, disorders of the peripheral nervous system, can also be confused with myoclonus. These phenomena all have classic physiologic findings on electromyography (EMG), allowing them to be distinguished from myoclonus (2).
CLASSIFICATION SCHEMES
Various schemes exist for classifying the large number of myoclonic disorders. Myoclonus can be classified on the basis of its clinical characteristics, its pathophysiology, its site of origin, or its underlying etiology (3–5).
Clinically, myoclonus can occur spontaneously, can be activated or accentuated by voluntary movement (action myoclonus), or can be activated or accentuated by external sensory stimulation (reflex myoclonus). It can be focal, involving only a few adjacent muscles, can be generalized, involving many or most of the muscles of the body, or can be multifocal, involving many muscles that jerk independently of one another. Myoclonic jerks can occur rhythmically, or can occur in an irregular fashion (2).
The starting point for physiologic classification is to classify myoclonus as epileptic or nonepileptic (6). The critical question is whether the myoclonus is an epileptic fragment. If epileptic, then this leads to symptomatic therapy with antiepileptic drugs. If not, the physiology needs further dissection to decide on symptomatic therapy.
Combining the clinical features of myoclonus with the results of electrophysiologic studies allows for more precise classification based upon the site of origin of the myoclonus (6–8). Cortical myoclonus originates in the sensorimotor cortex and is transmitted down the spinal cord in pyramidal pathways; it can develop into focal motor seizures with potential to secondarily generalize (9). Myoclonus originating from the brainstem may manifest as an exaggerated startle, or may manifest as generalized muscle jerks independent of startle mechanisms. Brainstem myoclonus may also take the form of palatal myoclonus (10), which is now often classified as palatal tremor. Myoclonus may also originate from the spinal cord, and take the form of spinal segmental myoclonus, which affects a few spinal segments, or propriospinal myoclonus, which produces generalized axial jerks that usually begin in the abdominal muscles (2). Rarely, local lesions of peripheral nerves, plexi, or nerve roots may produce peripheral myoclonus (11).
From an etiologic perspective, myoclonus can be considered physiologic, epileptic, essential, or symptomatic (2). Physiologic myoclonus refers to muscle jerks that occur in particular circumstances in normal individuals, such as sleep jerks (hypnic jerks) and hiccups. These are normal phenomena, and do not require treatment other than reassurance; these will not be further discussed in this chapter. In epileptic myoclonus, the epilepsy is predominant. Several of the major primary epileptic myoclonus syndromes are summarized in Table 31.1, including cryptogenic myoclonic epilepsy, juvenile myoclonic epilepsy, eyelid myoclonus with absence, infantile spasms, and Lennox–Gastaut syndrome (12–16). Of these, juvenile myoclonic epilepsy is the most frequent, typically presenting in adolescence (17). These will not be considered further in this chapter either. Essential myoclonus occurs in the absence of other significant neurologic deficit or abnormality, and is idiopathic or genetic. The myoclonus in symptomatic myoclonus is secondary to an underlying disorder; there is a long differential diagnosis, and the most common may be the dementing illnesses (1). We will consider these in detail below.
NEUROPHYSIOLOGIC ASSESSMENT
After a thorough physical examination, electrophysiologic assessment is the next step in evaluating the patient with myoclonus, and can help distinguish myoclonus from other hyperkinetic disorders as well as help to better characterize the myoclonus (6–8). Multichannel surface EMG, long-latency EMG responses to nerve stimulation, electroencephalography (EEG), EEG-EMG jerk-locked back averaging, and somatosensory-evoked potentials (SEP) are all useful tools in this assessment.
Surface EMG is useful in assessing the duration of EMG burst activity, and can be quite helpful in differentiating between epileptic and nonepileptic myoclonus. Epileptic myoclonus is typically characterized by EMG burst lengths ranging from 10 to 50 milliseconds (Fig. 31.1A), as well as synchronous antagonist muscle activity, and an EEG correlate. In contrast, nonepileptic myoclonus has longer EMG burst lengths of 50 to 300 milliseconds. Bursts in the 100-millisecond range are seen in essential myoclonus, whereas bursts of longer than 100 milliseconds are likely to be dystonic or psychogenic (Fig. 31.1B).
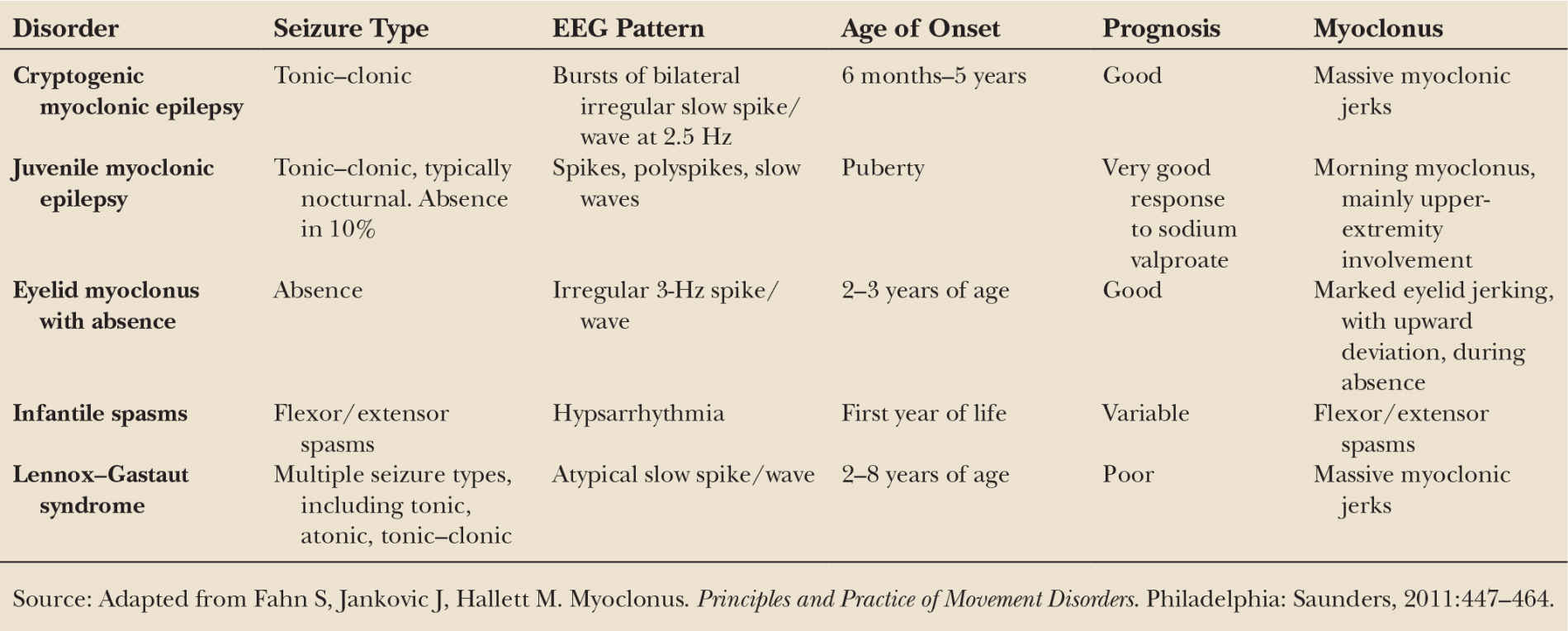
A brief lapse in muscle contraction of active postural muscles may result in a jerk that may appear visually indistinguishable from a movement produced by a burst of EMG activity (Fig. 31.1C). This is referred to as negative myoclonus, clinically called asterixis. Asterixis may be cortical or subcortical in origin; in the case of negative cortical reflex myoclonus, the giant somatosensory cortical potential is time-locked to absence of EMG activity (18,19).
Surface EMG also helps determine the distribution of muscle involvement, further assisting with localization of the origin of the myoclonus. Lesions of a peripheral nerve, part of a plexus, a spinal root, or a restricted number of spinal cord segments would be expected to give rise to a characteristic pattern of muscle involvement. Myoclonic jerks involving axial muscles, including the neck, shoulders, trunk, and hips, may arise in the brainstem as an exaggerated startle response or as brainstem reticular myoclonus, or in the spinal cord as propriospinal myoclonus. Brainstem myoclonus can be differentiated from propriospinal myoclonus by pattern of muscle involvement. In brainstem myoclonus, myoclonus is initially activated in cranial nerve muscles from the XI nerve nucleus up the brainstem, with limb and axial muscles activated in descending order. In contrast, propriospinal myoclonus is initially activated in muscles innervated by the thoracic cord, with slow upward and downward spread (6).
Electrophysiology can also be useful in identifying psychogenic myoclonus. In particular, the presence of a Bereitschaftspotential beginning approximately one second prior to the EMG burst on back-averaged EEG, as is seen in voluntary movement, supports the diagnosis of psychogenic myoclonus (20,21) (Fig. 31.2A). Stimulus-evoked myoclonus that is of variable latency and longer than a voluntary reaction time is also suggestive of psychogenic myoclonus (21–23).
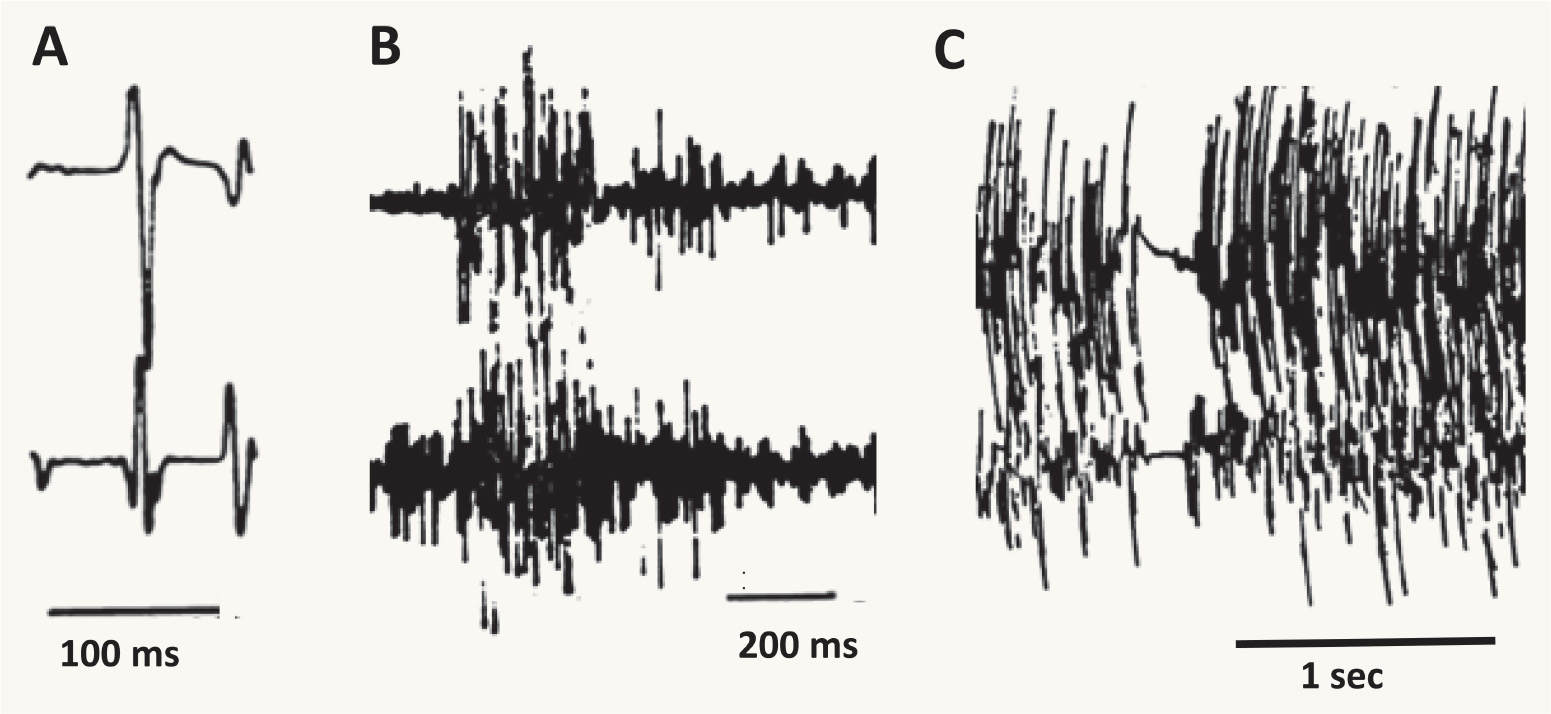
Figure 31.1 EMG patterns underlying three different types of myoclonus. A is from a patient with epileptic myoclonus, B is from a patient with dystonic myoclonus, and C is from a patient with negative myoclonus. The time scale is noted for each. Note that in A the EMG burst is very short, while in B the EMG burst is considerably longer. Note the transient lapse in EMG activity in C. (Adapted courtesy of the Hallett lab.)
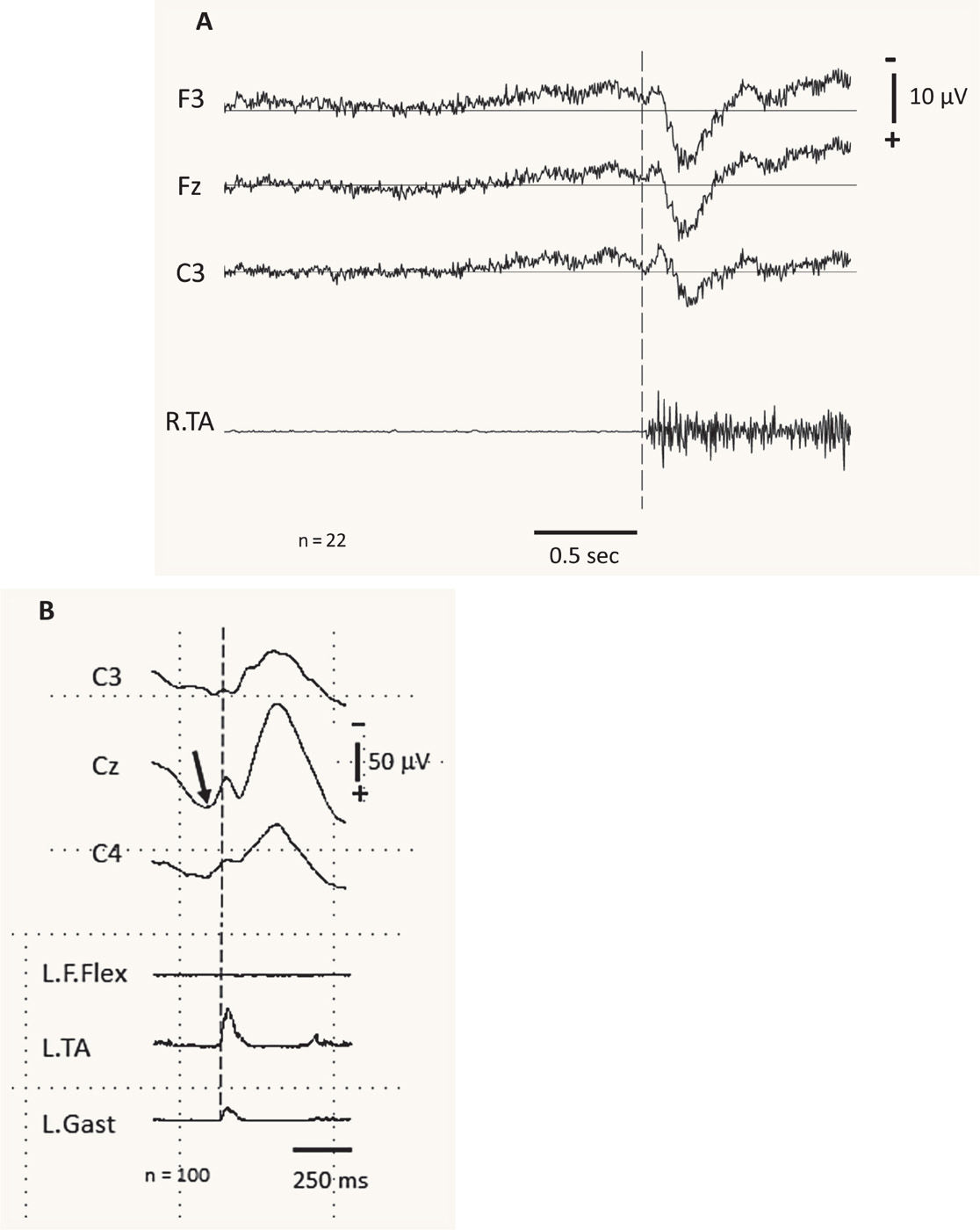
Figure 31.2 EMG-EEG jerk-locked back averaging observed in different types of myoclonus. A is from a patient with functional (psychogenic) myoclonus, and B is from a patient with cortical myoclonus. A demonstrates the average of 22 EEG and EMG events associated with involuntary myoclonic jerks in a patient with functional myoclonus. The top three traces are average EEG records from the left frontal (F3), frontal midline (Fz), and left central (C3) electrodes, referenced to linked earlobes. The bottom trace is the EMG from the right tibialis anterior (R.TA). Note the presence of a Bereitschaftspotential beginning approximately 1 second prior to the EMG burst on back-averaged EEG. B demonstrates the average of 100 EEG and EMG events associated with spontaneous cortical myoclonus. Note the focal cortical activity (arrow) at the central midline electrode (Cz) immediately prior to the EMG activity noted in the left tibialis anterior (L.TA). (A courtesy of R. Paine; B adapted courtesy of the Hallett lab.)
PHYSIOLOGICALLY CHARACTERIZED EPILEPTIC MYOCLONUS
Electrophysiologic studies can further subdivide epileptic myoclonus into three types: cortical myoclonus, reticular myoclonus, and primary generalized epileptic myoclonus (6,7).
Cortical myoclonus is characterized by short-duration EMG bursts, arrhythmicity, rapid craniocaudal recruitment, increased sensitivity to tactile stimuli, and predominant distribution in the face and distal extremities. Abnormal focal discharges in the sensorimotor cortex contralateral to the myoclonic jerk can be recorded from EEG-EMG jerk-locked back averaging (6,8) (Fig. 31.2B). Cortical myoclonus may be characterized by the presence of pathologically enlarged SEPs. Peripheral stimuli generating giant SEPs often instigate a subsequent myoclonic burst (the C reflex) at a latency compatible with activation through fast-conducting corticomotoneuron pathways. Subcortical myoclonus, on the other hand, is suggested when the reflex myoclonus triggered by peripheral stimuli occurs with a latency too short to involve cortical pathways (24,25).
Cortical myoclonus can be further classified into spontaneous cortical myoclonus, cortical reflex myoclonus, and epilepsia partialis continua. Muscle jerks can occur spontaneously (spontaneous cortical myoclonus), or can be triggered by external stimuli (cortical reflex myoclonus) or by movement (cortical action myoclonus) (9,26). Cortical myoclonus may be focal or multifocal, depending upon whether it results from a single or from multiple discrete discharges in the motor cortex. Cortical discharges can also result in generalized muscle jerks via intracortical or transcallosal spread (27,28). Increased cortical excitability may be secondary to loss of inhibitory interneurons (29). Epilepsia partialis continua, a subtype of cortical myoclonus, can be defined as the continuous focal jerking of a body part, occurring over hours, days, or years, secondary to a cerebral cortical abnormality (30). Focal cortical myoclonus can result from almost any type of focal cortical lesion, including tumors, encephalitis, angiomas, or focal cortical dysplasias. Surgical excision of excitable tissue should be considered, as it has been demonstrated to be curative in some cases.
Reticular myoclonus is observed in some types of generalized epilepsy. It is characterized by a generalized, mainly axial, myoclonic jerk with neck flexion, shoulder elevation, and trunk and knee extension. This type of myoclonus originates in muscles innervated by the caudal brainstem, then spreads up the brainstem and down the spinal cord (31). Electrophysiologically, this type of myoclonus is characterized by brief generalized EMG bursts lasting 10 to 30 milliseconds, triggered by sensory stimulation or action. EMG activation begins with the sternocleidomastoid muscle, and other cranial nerve muscles are subsequently activated in reverse numerical order. Reticular myoclonus lacks a time-locked EEG correlate. It may occur after cerebral anoxia, with renal failure, and can also be seen in stiff-man syndrome (32) as well as with brainstem lesions as occur in multiple sclerosis (33).
Primary generalized epileptic myoclonus refers to a set of disorders characterized predominantly by brief myoclonic, atonic, or tonic seizures. Patients with these disorders may also experience massive myoclonic jerks that can represent either positive or negative myoclonus (34–36).
In terms of therapy, the use of anticonvulsant drugs is generally first line for symptomatic treatment of epileptic myoclonus. Agents that enhance GABA action appear to be especially helpful. Unlike the treatment of seizures, where monotherapy is often possible, patients with myoclonus often require multiple agents to control their movements. The doses of various commonly used agents are listed in Table 31.2. Epileptic myoclonus often responds best to sodium valproate and clonazepam used in combination at maximum tolerated anticonvulsant doses (37). If disability remains, piracetam or levetiracetam can be added (38–40). Phenytoin, carbamazepine, and gabapentin should be avoided, as they may worsen myoclonus.
Treatment of negative myoclonus should initially entail searching for an underlying metabolic or toxic cause. Negative myoclonus is frequently refractory to medication, although levetiracetam (41,42) has been demonstrated to be effective at treating epileptic negative myoclonus.
SYMPTOMATIC MYOCLONUS
Symptomatic myoclonias constitute an appreciably heterogeneous group. Based upon physiologic criteria, myoclonias in this group may be epileptic or nonepileptic. Anticonvulsant medications may or may not be useful in this group of disorders; treatment directed toward the underlying cause should be most helpful if possible.
| Appropriate Dosages of Agents for the Treatment of Myoclonus |
Drug | Dosage (mg/d) |
Baclofen | 15–100 |
Benztropine | 4–9 |
Clonazepam | 3–20 |
Diazepam | 5–30 |
5-Hydroxytryptophan | Up to 1,500* |
Levetiracetam | 1,000–3,000 |
Phenobarbital | 60–180 |
Piracetam | 2,400–16,800 |
Primidone | 500–750 |
Tetrabenazine | 50–200 |
Trihexyphenidyl (benzhexol) | Up to 35 |
Valproic acid (sodium valproate) | 1,000–2,000 |
Zonisamide | 100 to 600 |
*In combination with a peripheral aromatic amino acid decarboxylase inhibitor (such as carbidopa 100 to 300 mg/d). Source: Adapted from Fahn S, Jankovic J, Hallett M. Myoclonus. Principles and Practice of Movement Disorders. Philadelphia: Saunders, 2011:447–464. | |
MYOCLONUS AS A FRAGMENT OF OTHER INVOLUNTARY MOVEMENT DISORDERS
Patients with disorders including Wilson’s disease, neuroaxonal dystrophy, pantothenate kinase–associated neurodegeneration (PKAN), progressive supranuclear palsy, and parkinsonism may all manifest myoclonus. Myoclonus can also be seen in Huntington’s disease, and can respond to treatment with valproic acid (43).
Myoclonus is rare in Parkinson’s disease. When present, it is most frequently the result of dopa-dyskinesias (44), and can be treated by regulating the dose of levodopa, by medications such as amantadine, or by deep brain stimulation. Patients with Parkinson’s disease may also have orthostatic myoclonus while standing that may contribute to balance impairment (45,46). Myoclonus is more common in the Parkinson’s plus syndromes multiple system atrophy (47) and corticobasal degeneration (48). Corticobasal degeneration in particular is characterized by stimulus-sensitive myoclonus with a short latency.
NEURODEGENERATIVE—THE PROGRESSIVE MYOCLONUS EPILEPSIES
In the progressive myoclonus epilepsies, severe myoclonus (spontaneous, action, and stimulus-sensitive) and severe seizures are accompanied by slowly progressive neurologic decline, typically involving dementia and ataxia (49–54). Symptoms typically begin between 7 and 15 years of age, and syndromes are quite heterogeneous. While there is no cure for these neurodegenerative conditions, it is of critical importance to recognize them, and avoid treatment with phenytoin, which can result in significant clinical worsening (55). The differential diagnosis of progressive myoclonus epilepsy is briefly discussed below.
Unverricht–Lundborg disease (EPM1) is characterized by stimulus-sensitive myoclonus, tonic–clonic seizures, progressive course with ataxia but only mild intellectual decline, and a characteristic EEG pattern, with paroxysmal generalized sharp-wave activity and photosensitivity (56,57); age of onset is between 6 and 15 years of age. It is inherited in an autosomal recessive manner, and in a number of families, the disorder has been linked to a mutation in cystatin B, a small protein in the superfamily of cysteine protease inhibitors (58). While the exact role of cystatin B is unknown, it interacts with a variety of proteins found in the cerebellum (59), and may be involved in sensitization to oxidative stress (60).
Lafora body disease (EPM2) is an autosomal recessive, adolescent-onset, progressive, and fatal generalized mucopolysaccharide storage disorder. The predominant central nervous system manifestations include stimulus-sensitive myoclonias, seizures, behavioral changes, dementia, and ataxia. Dementia is present in two-thirds of patients within 2 years after onset, and in all patients within 5 years. Death occurs in 80% by 18 years of age (61). Polyglucosan acid–Schiff (PAS)-positive inclusion bodies are found throughout the cerebral and cerebellar gray matter, in addition to being found in liver, muscle, and skin (62,63). Approximately 75% of cases of Lafora body disease are attributed to mutations in the gene laforin (EPM2A), which encodes for a protein tyrosine phosphatase (64). Mutations in the gene malin (EPM2B, or NHLRC1), which encodes for a putative E3 ubiquitin ligase, are also associated with Lafora body disease (65). Laforin and malin bind to one another (66), and may regulate cellular glucose uptake by preventing plasma membrane targeting of glucose transporters (67). In terms of treatment, zonisamide has been demonstrated to be useful in treating the myoclonus and epilepsy in patients with Lafora body disease (68).
Neuronal ceroid lipofuscinosis (Batten disease) is a heterogeneous autosomal recessive disorder with late-infantile, juvenile, and adult-onset forms. In addition to presenting with seizures, myoclonus, and cognitive impairment, it also presents with blindness in the late-infantile and juvenile forms (57,69,70
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


