Benign Myoclonic Epilepsy in Infancy
Charlotte Dravet*
Michelle Bureau*
*Centre Saint-Paul-Hôpital Henri Gastaut, Marseille, France
Introduction
The syndrome of benign myoclonic epilepsy in infancy (BMEI) was not clearly identified before the first description in seven infants in 1981 (1). In this article, it was defined as the occurrence of myoclonic seizures (MS) without other seizure types, except rare simple febrile seizures (FS), in the first 3 years of life in normal infants. These MS were easily controlled by a simple treatment and remitted during childhood. The psychomotor development remained normal and no severe psychologic consequences were observed. Many other cases have been published since. BMEI was classified among the generalized idiopathic epilepsies in the1989 international classification (2). Several authors have described cases with reflex MS, triggered by noise or contact, and have proposed distinguishing two separate entities, the second one being named Reflex myoclonic epilepsy in infancy (3). We do not think this distinction is necessary, and we will describe all the cases as BMEI.
To our knowledge, there are, at present, 115 cases published in the literature, of which 95 correspond to the classic description (4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21) and 20 were reported as reflex BMEI (3,22,23,24,25,26,27,28). We now have five new cases, of which four have associated spontaneous and reflex seizures, making a total of 120. One also must mention that in the first description, the onset was before 3 years of age, whereas in the following reports some cases have a later onset, up to 4 years 8 months (15). That means the same type of epilepsy may appear at different ages, but tends to be more frequent in some periods (29).
General Background
Epidemiology
According to the few available epidemiological data, BMEI seems to represent less than 1% of all the epilepsies (30 and unpublished data from the Centre Saint-Paul, 1999), 2% of all idiopathic generalized epilepsies (unpublished data from the Centre Saint-Paul, 1997), 1.3%, and 1.72% of epilepsies, which begin in the first year of life (14,19), around 2% of epilepsies, which begin in the first 3 years of life (4), and 0.39% of epilepsies which begin in the first 6 years of life (12).
Sex
When the sex is mentioned, it appears that boys outnumber girls: 62 vs. 34.
Genetics
Genetics of BMEI are unknown. Cases are rare and no family cases of BMEI have been described. A family history of epilepsy or febrile seizures (FS) was present in 44% of 88 cases. In 78 cases, the rate of FS in the family was 17% and the rate of epilepsy 27%. The epilepsy type found in relatives is difficult to assess. In ten cases, it was probably an idiopathic epilepsy. In the case described by Arzimanoglou et al. (13) the proband was the second of two brothers and the oldest was affected by a typical epilepsy with myoclonic-astatic seizures (EMAS, Doose syndrome). In the family of the patient described by Biondi et al. (8), other members had dubious epilepsy. Sleep EEGs were obtained for the father and his two sisters and demonstrated brief bursts of generalized spike-waves. In one of our patients (unpublished case), the maternal aunt suffered from myoclonic jerks at adolescence, but the diagnosis of juvenile myoclonic epilepsy was not established.
Personal History
Most patients do not have any pathologic history prior to the onset of the MS. Only two (1.9%) had an associated disease: Down’s syndrome (10) and hyperinsulinic diabetes (5). One bleeding during pregnancy (15) and one cesarean birth (22) were mentioned, and Badinand-Hubert et al. (7), indicated that three patients presented with “neurological or neuropsychological syndromes” without precision.
However, the occurrence of FS is not uncommon: 20 patients among 81 (25%). They were always simple, usually rare (one or two), and were observed before the onset of the myoclonias and before initiation of treatment (15 cases). One single study (17) reported the description by the parents of infrequent, generalized afebrile seizures, at the early onset or during drug withdrawal, never observed by professionals. The authors believed they were repeated MS.
Clinical and Electroencephalogram Manifestations
The age at onset is usually between 4 months and 3 years. An earlier onset is uncommon. A later onset was reported, at 4 years 9 months (15) and at 3 years 9 months (17).
Initially, the MS are brief, often rare, involving the upper limbs and the head, and rarely the lower limbs. In babies, they may be barely noticeable and the parents sometimes have difficulty in determining their exact onset and their frequency. They often speak of “spasms” or “head nodding.” Later on, their frequency increases.
Video-EEG and polygraphic recordings have enabled us to make a precise analysis of these seizures. They are more or less massive myoclonic jerks, involving the axis of the body and the limbs, provoking a head drop and an upward-outward movement of the upper limbs, with flexion of the lower limbs, and, sometimes, a rolling of the eyeballs. Their intensity varies from one child to the other and from one attack to another in the same child. The most severe forms cause a sudden projection of the objects held in the hands, and, sometimes, a fall. The mildest forms provoke only brief forward movement of the head, or, even, a simple closure of the eyes. As a rule, the seizures are very brief (1–3 s), although they may be longer, especially in older children, consisting of pseudorhythmically repeated jerks lasting no more than 5 to 10 s. They occur several times a day at irregular and unpredictable times. Unlike infantile spasms, they do not occur in long series. They are not favored by awakening, but rather by drowsiness. In some patients, they can be triggered by intermittent photic stimulation (IPS). In the patients with reflex BMEI, they are triggered by a sudden noise or a sudden contact. The state of consciousness is difficult to assess in isolated seizures. Only when they are repeated is there a slight impairment of consciousness without interruption of activity. The patient reported by Zafeiriou et al. (28) presented reflex myoclonic seizures of a longer duration (up to 5 s) with eyelid myoclonias and an absence component. This seizure type was also reported by Prats-Vinas et al. (21) in three of their eight patients. We never observed the sudden brief vocalization reported by Lin et al. (17), ascribed by the authors to the involvement of the diaphragm and/or the abdominal muscles producing an expiratory noise. In reflex BMEI, the myoclonus is elicitable both in wakefulness and in sleep, with a threshold lower in stage I and increasing gradually during the slower stages (3). No REM sleep was recorded and tested in patients with reflex MS.
As the development continues normally, parents and pediatricians tend not to consider these movements as pathologic events.
When an EEG is performed, it can be normal on awakening if no myoclonic fit is recorded. However, myoclonias are always associated with an EEG discharge. Polygraphic recordings demonstrate that the myoclonias are accompanied by a discharge of fast generalized spike-waves or polyspike-waves at more than 3 Hz, lasting the same time as the myoclonias (Fig. 9-1). This discharge is more or less regular and can start in the two anterior areas and the vertex. Myoclonias are brief (1–3 s) and usually isolated. The myoclonic jerk may be followed by a brief atonia, sometimes, after the attack. There is a voluntary movement, visible as a normal muscular contraction. In only one patient did we observe the association of myoclonias in the deltoid muscle with a pure atonia in the neck muscles. During drowsiness, there is an enhancement of the myoclonias; they usually, but not always, disappear during slow sleep. The MS triggered by tactile and acoustic stimuli have the same characteristics. Ricci et al. (3) noted that the initial manifestation generally, but not always, consisted of a blink, followed 40–80 msec later by the first myoclonic arm jerk. After a myoclonic attack there was a refractory period, lasting 20 to 30 s to 1 to 2 min, during which sudden stimuli did not provoke attacks, even when the startle reaction was easily elicited. IPS can also provoke MS (10,11,15,17,21).
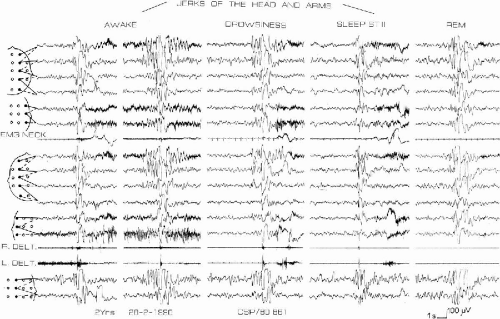 FIG. 9-1. In a 2-year-old girl, before any treatment, several myoclonic jerks are polygraphically recorded when awake, persisting during drowsiness, and attenuated during sleep, stage II. They are accompanied by generalized spike-wave, sometimes preceded by spike-wave localized in the anterior regions. The same type of generalized discharge appears during REM without concomitant clinical event. R DELT: right deltoid muscle; L DELT: left deltoid muscle. (From Dravet C, Bureau M, Roger J. Benign myoclonic epilepsy in infants. In: Roger J, Bureau M, Dravet C, Dreifuss FE, Perret A, Wolf P, eds. Epileptic syndromes in infancy, childhood and adolescence, 2nd ed. London, Paris: John Libbey, 1992:67–74. With permission.) |
The interictal EEG is normal for the child’s age. Spontaneous spike-wave discharges are rare; some slow waves may be found over the central areas. Surprisingly, the patient with reflex BMEI reported by Kurian and King (27) had an awake EEG with numerous discharges of spike-waves and polyspike-waves without clinical correlate. IPS does not provoke spike-waves without concomitant myoclonia at the onset. Nap sleep recordings have shown a normal organization of sleep; generalized spike-wave discharges may occur during rapid eye movement (REM) sleep (Fig. 9-1).
Evolution and treatment
No other type of seizure is observed in children with BMEI, even if they are left untreated (for up to 8.5 years in one of our patients), particularly no absence or tonic seizure. Clinical examination is normal. Interictal myoclonus was described only by one author (15) in six patients. By reviewing our own patients, we found a mild interictal myoclonus in two, revealed by polygraphic recordings.
Many patients were not investigated, but when CT scan and MRI were performed they were normal (43 patients).
The outcome seems to depend on an early diagnosis and treatment. Myoclonias are easily controlled by Valproate (VPA) alone and the child may then develop regularly, according to the normal milestones. If left untreated, the patient continues to experience myoclonic attacks and this may lead to impaired psychomotor development and behavior disturbances.
The therapeutic modalities are known more or less precisely in 91 patients. It consisted of a monotherapy in 81 patients, polytherapy in 7, and no therapy in 3. Monotherapies were with VPA in 76, with phenobarbital (PB) in 2, with clobazam (CLB) in 2, and with nitrazepam (NTZ) in 1. Seventy one (88%) became seizure-free (67 with VPA, 2 with PB, and 2 with CLB). Nine patients with VPA had a seizure reduction, of which four became seizure-free after addition of PB, clonazepam (CZP) or CLB (15). We do not know the evolution for the five others. One patient with NTZ also had a seizure reduction, followed by a complete control after addition of VPA. Six patients (11) received bitherapy with primidone (PRM), and ethosuximide (ESM), of which only one became seizure-free. In the other five, the addition of VPA followed by VPA monotherapy allowed complete control. Only in one patient did myoclonic seizures persist, in spite of various polytherapies and even a ketogenic diet (21). Finally, three patients presenting with reflex seizures did not receive any treatment and became seizure-free spontaneously (3). On the whole, 85 patients (93%) became seizure free.
These data confirm that VPA is the drug of first choice in BMEI. However, the treatment must be monitored using plasma-level assessments because an irregular intake can lead to a relapse and falsely mimic a drug-resistant epilepsy. Lin et al. (17) gave a very detailed study of the treatment in their patients. They also underlined the necessity of this monitoring and of the use of high doses at the onset (30–40 mg/k) to obtain levels more than 100 mg/l, 3 hours after the morning intake, in some patients. The daily dose was reduced to usual therapeutic plasma levels (50–100 mg/l) after seizures were controlled.
Related posts:
 Myoclonic Status in Nonprogressive Encephalopathies
Severe Myoclonic Epilepsy in Infancy: Dravet Syndrome
Eyelid Myoclonia and Absence
Childhood Absence Epilepsy Evolving to Juvenile Myoclonic Epilepsy: Electroclinical and Genetic Features
Familial Adult Myoclonic Epilepsy (FAME)
Treatment of Myoclonic Epilepsies in Infancy and Early Childhood
Myoclonic Status in Nonprogressive Encephalopathies
Severe Myoclonic Epilepsy in Infancy: Dravet Syndrome
Eyelid Myoclonia and Absence
Childhood Absence Epilepsy Evolving to Juvenile Myoclonic Epilepsy: Electroclinical and Genetic Features
Familial Adult Myoclonic Epilepsy (FAME)
Treatment of Myoclonic Epilepsies in Infancy and Early Childhood
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


