(22)—although less effective than full agonists like diazepam, are anticonvulsant in animal models and appear less likely to promote tolerance (23,24). Other “inverse agonists” at the benzodiazepine site, including some β-carbolines, inhibit GABA binding or GABA-evoked currents (25). They can induce convulsive seizures or anxiety (25,26) but as yet have no clinical utility.
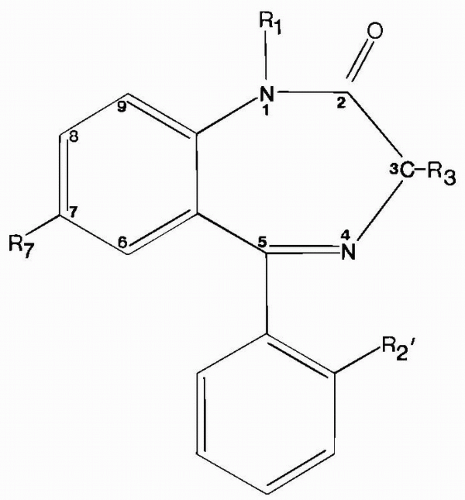 Figure 58.1 1,4-Benzodiazepine structure. For anticonvulsants, R1 = H or CH3; R3 = H, OH, or COO−; R7 = Cl or NO2; and R2′ = H or Cl. |
TABLE 58.1 ANTICONVULSANT ACTIVITY, MOTOR IMPAIRMENT, AND RECEPTOR BINDING OF SOME BENZODIAZEPINES | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||
for GABA leftward (Fig. 58.2), increasing current amplitudes at lower GABA concentrations but not increasing maximal current (38). This shift is caused by an enhanced affinity for GABA at its binding site, with no change in channel-gating kinetics (37). The benzodiazepines thus increase the current produced by low GABA concentrations but have no effect at high GABA concentrations, at which receptor binding is saturated. Studies of GABAergic inhibitory postsynaptic currents (IPSCs) have suggested that GABA is present in the synaptic cleft for 1 to 3 ms at high concentrations (about 1 mM) (39,40). Thus, at individual synapses, benzodiazepines prolong the mIPSC decay phase (41,42) by slowing the dissociation of GABA from the receptor (43,44) without changing maximal mIPSP amplitude. Prolongation of the mIPSC increases the likelihood of temporal and spatial summation of multiple synaptic inputs, which, in turn, raises the amplitude of stimulus-evoked IPSCs. The benzodiazepines thus increase the inhibitory “tone” of GABAergic synapses, which prevents or limits the hypersynchronous firing of neuron populations that underlies seizure activity (45).
removed from patients with temporal lobe epilepsy (76). Changes in the GABA current reversal potential might also explain why diazepam can be less effective in children with epileptic encephalopathies (77), and why, rarely, it can cause status epilepticus in patients with Lennox-Gastaut syndrome (78,79).
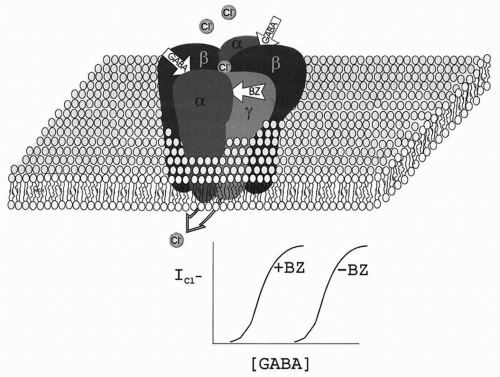 Figure 58.2 Model of a GABAA receptor in the plasma membrane. The receptor consists of five closely related subunits, each with four membrane-spanning domains. The receptor is a ligand-gated anion channel, with two binding sites for GABA (between α and β subunits) and one for benzodiazepines (between α and β subunits). In the presence of GABA, the channel becomes permeable to chloride ions, producing the fast inhibitory postsynaptic potential (IPSP). In the presence of a benzodiazepine agonist, GABA binds more efficiently, enhancing the IPSP. Benzodiazepines have no effect in the absence of GABA. |
Pharmacokinetic interactions with other anticonvulsants except phenobarbital are infrequent and inconsistent. Diazepam enhances phenobarbital elimination (167), and phenobarbital increases the clearance (168) and lowers plasma levels of clonazepam (169). Valproate reduces diazepam protein binding, increasing free drug levels (170), and enhances diazepam’s CNS effects (167). Other AEDs may augment metabolism and clearance of N-desmethyldiazepam derived from clorazepate (171). Clobazam increases the 10-11 epoxide metabolite of carbamazepine (172).
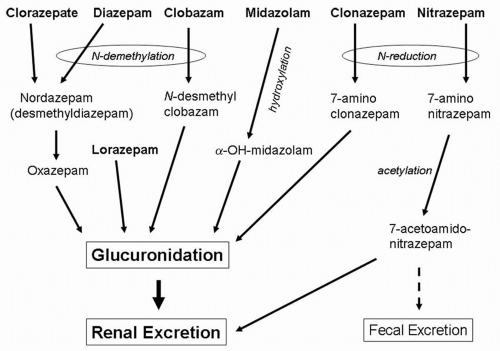 Figure 58.3 Metabolism of the anticonvulsant benzodiazepines. |
TABLE 58.2 CLINICAL PHARMACOLOGY OF BENZODIAZEPINES USED FOR ACUTE SEIZURES | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
of efficacy for specific indications with the individual agents are discussed below.
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


