Valproate
Blaise F. D. Bourgeois
HISTORICAL BACKGROUND
The anticonvulsant effect of valproic acid, or valproate (VPA), was discovered serendipitously when the agent was used as a solvent for compounds tested in an animal model of seizures (1). VPA has been used in the treatment of epilepsy for more than 30 years (2). In the United States, VPA was approved in 1978. Since then, it has been regarded as one of the major antiepileptic drugs (AEDs), distinguished from previous agents by its broad spectrum of activity against many seizure types as well as by its relatively low sedative effect. In addition to being the first agent to be highly effective against several primarily generalized seizure types, such as absences, myoclonic seizures, and tonic-clonic seizures, VPA was found to be effective in the treatment of partial seizures, Lennox-Gastaut syndrome, infantile spasms, neonatal seizures, and febrile seizures. Although VPA is also used in the treatment of affective disorders, migraine headaches, and Sydenham chorea (3), these indications will not be included in the present discussion.
CHEMISTRY AND MECHANISM OF ACTION
Valproate (MW 144.21; Fig. 54.1), a short-chain, branched fatty acid, is a colorless liquid with low solubility in water. Other forms include: (a) sodium valproate (MW 166.19), a highly water-soluble and highly hygroscopic white, crystalline material; (b) sodium hydrogen divalproate (divalproex sodium), a complex composed of equal parts of VPA and sodium valproate (Fig. 54.1); and (c) magnesium valproate, a divalproate salt. The antiepileptic activity of VPA, demonstrated in several animal models (4,5), includes protection against maximal electroshock-induced seizures; against seizures induced chemically by pentylenetetrazol, bicuculline, glutamic acid, kainic acid, strychnine, ouabain, nicotine, and intramuscular penicillin; and against seizures induced by kindling (6). This broad spectrum of efficacy of VPA in animal models suggests that the agent is effective in both preventing the spread and lowering the threshold of seizures. Although several effects of VPA have been demonstrated at the cellular level, the precise mechanism underlying the antiepileptic effect of the agent has not been identified, and more than one mechanism may be involved. Identified mechanisms include potentiation of γ-aminobutyric acid (GABA)ergic function, inhibition of γ-hydroxybutyric acid formation, reduction of excitation mediated by glutamate receptors, and inhibition of voltagesensitive sodium channels (7). It is not known to what extent any of these actions contributes to clinical seizure protection by VPA.
ABSORPTION, DISTRIBUTION, AND METABOLISM
The main pharmacokinetic parameters of VPA are summarized in Chapter 45, Table 45.2. Different preparations of VPA are available, although not all are available in any given country. Oral preparations of VPA include VPA capsules, tablets, and syrup (immediate-release); enteric-coated tablets of sodium valproate or sodium hydrogen divalproate (divalproex sodium); divalproex sodium enteric-coated sprinkles; slow-release oral preparations; magnesium valproate; and valpromide (the amide of VPA). VPA suppositories and a parenteral formulation of sodium valproate for intravenous (IV) use are also available. The bioavailability of oral preparations of VPA is virtually complete compared with that of the IV route (8). The purpose of the enteric coating of tablets is to prevent gastric irritation associated with release of VPA in the stomach. Compared with oral syrup, the relative bioavailability of VPA suppositories was found to be 80% in volunteers (9). In a study of patients treated chronically with VPA, administration of VPA suppositories was well tolerated for several days, and the bioavailability was the same as that of the oral preparations (10).
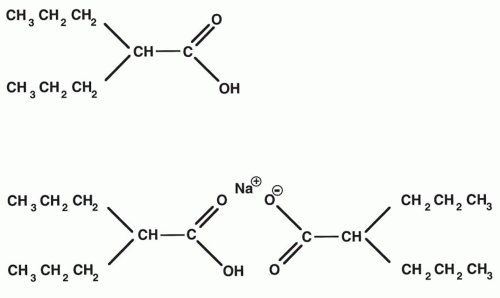 Figure 54.1 Structural formulas for valproic acid (N-dipropylacetic acid) and sodium hydrogen divalproate (divalproex sodium). |
The rate of absorption of VPA after oral administration is variable, depending on the formulation. Administration of syrup or uncoated regular tablets or capsules is followed by rapid absorption and peak levels within 2 hours. Absorption from enteric-coated tablets is delayed but rapid. The onset of absorption varies as a function of the state of gastric emptying at the time of ingestion, and peak levels may be reached only 3 to 8 hours after oral ingestion of enteric-coated tablets (11, 12, 13; Chapter 45, Fig. 45.1). Therefore, in patients treated chronically with enteric-coated VPA, the true trough level may occur in the late morning or early afternoon (10). The bioavailability of enteric-coated sprinkles of divalproex sodium was compared with that of VPA syrup in 12 children, with no difference noted between the two formulations (14). However, the average time to maximal VPA concentrations was longer for sprinkles (4.2 hours) than for syrup (0.9 hours). With the IV formulation, peak VPA serum levels are reached at the end of the recommended infusion time of 60 minutes.
The volume of distribution of VPA is relatively small (0.13 to 0.19 L/kg in adults and 0.20 to 0.30 L/kg in children). VPA is highly bound to serum proteins; this binding appears to be saturable at therapeutic concentrations, with the free fraction of VPA increasing as the total concentration increases (15): 7% at 50 mg/L, 9% at 75 mg/L, 15% at 100 mg/L, 22% at 125 mg/L, and 30% at 150 mg/L. On the basis of these values, with an only three-fold increase in the total concentration of VPA, from 50 to 150 mg/L, the free level of VPA would increase more than 10 times, from 3.5 to 45 mg/L. Accordingly, a curvilinear relationship between VPA maintenance dose and total steady-state concentrations was found, with relatively smaller increases in concentrations at higher doses (16). The elimination half-life of VPA varies as a function of comedication. In the absence of inducing drugs, the half-life in adults is 13 to 16 hours (11,17), whereas in adults receiving polytherapy with inducing drugs, the average half-life is 9 hours (8). In children, the half-life is slightly shorter. Cloyd and colleagues (18) reported an average half-life of 11.6 hours in children receiving monotherapy and 7.0 hours in those receiving polytherapy. Newborns eliminate VPA slowly; the half-life in this population is longer than 20 hours (19).
The most abundant metabolites of VPA are glucuronide and 3-oxo-VPA, which represent about 40% and 33%, respectively, of the urinary excretion of a VPA dose (20). Two desaturated metabolites of VPA, 2-ene-VPA and 4-ene-VPA, have anticonvulsant activity that is similar in potency to that of VPA itself (21). Because there is delayed but significant accumulation of 2-ene-VPA in the brain and because it is cleared more slowly than VPA (22), the formation of 2-ene-VPA provides a possible explanation for the discrepancy between the time courses of VPA concentrations and antiepileptic activity (23). It appears that 2-ene-VPA does not have the pronounced embryotoxicity (24) and hepatotoxicity (25) of 4-ene-VPA. Both are produced by the action of cytochrome P450 enzymes, which are induced by certain other AEDs (20,26). This may explain the increased risk for hepatotoxicity in patients receiving VPA concomitantly with these agents (27). However, elevation of 4-ene-VPA levels has not yet been found in patients with VPA hepatotoxicity, short-term adverse effects, or hyperammonemia (28).
DRUG INTERACTIONS
Pharmacokinetic interactions with VPA fall into three categories, based on the following features: (a) the metabolism of VPA is sensitive to enzymatic induction; (b) VPA itself can inhibit the metabolism of other agents; and (c) VPA has a high affinity for serum proteins and can displace other agents or be displaced from proteins (29, 30, 31). Concomitant administration of enzyme-inducing drugs has been repeatedly shown to lower VPA levels relative to the dose (32). Carbamazepine (33, 34, 35) and phenytoin (35) lower VPA levels by one-third to one-half, or even
more, in children (36, 37, 38). When children receiving polytherapy discontinued treatment with other agents, VPA levels increased 122% after withdrawal of phenytoin, 67% after withdrawal of phenobarbital, and 50% after withdrawal of carbamazepine (39). In contrast, levels of VPA are increased by coadministration of felbamate: 28% with felbamate 1200 mg per day and 54% with felbamate 2400 mg per day (40,41). According to one study, clobazam may significantly reduce the clearance of VPA (42).
more, in children (36, 37, 38). When children receiving polytherapy discontinued treatment with other agents, VPA levels increased 122% after withdrawal of phenytoin, 67% after withdrawal of phenobarbital, and 50% after withdrawal of carbamazepine (39). In contrast, levels of VPA are increased by coadministration of felbamate: 28% with felbamate 1200 mg per day and 54% with felbamate 2400 mg per day (40,41). According to one study, clobazam may significantly reduce the clearance of VPA (42).
VPA affects the kinetics of other drugs either by enzymatic inhibition or by displacement from serum proteins. Phenobarbital levels have been found to increase by 57% (43) to 81% (44) after the addition of VPA. Levels of ethosuximide can also be raised by the addition of VPA, mostly in the presence of additional AEDs (45). Although VPA does not increase levels of carbamazepine itself, levels of the active metabolite carbamazepine-10,11-epoxide may double (46,47). Elimination of lamotrigine is markedly inhibited by VPA, resulting in a two-to three-fold prolongation of the lamotrigine half-life (48). Although this is a competitive interaction that is likely to be rapidly reversible upon discontinuation of VPA, the inhibition seems to persist even at low VPA concentrations (49,50). A pharmacokinetic interaction occurs between VPA and phenytoin, because both agents have a high affinity for serum proteins. VPA increases the free fraction of phenytoin (51,52). Thus, in the presence of VPA, total phenytoin concentrations in the usual therapeutic range may be associated with clinical toxicity. In contrast to inducing AEDs, VPA is not associated with oral contraceptive failure (53).
EFFICACY
Very early it became apparent that VPA is a highly effective first-line agent for the treatment of primarily generalized idiopathic seizures, such as absence seizures, generalized tonic-clonic seizures, and myoclonic seizures (54). The indication for VPA when it was first released in North America in 1978 was for treatment of absence seizures. In patients with typical and atypical absence seizures, a reduction of spike-and-wave discharges was repeatedly demonstrated (55, 56, 57, 58). In two studies, comparison of VPA and ethosuximide for the treatment of absence seizures showed equal efficacy for the two agents (59,60). It appears that absence seizures are more likely to be fully controlled when they occur alone than when they are mixed with another seizure type (61,39). Overall, VPA appears to be somewhat less effective against atypical or “complex” absences than against simple absences (62,63). VPA can also be used effectively in patients with recurrent absence status (64).
VPA was found to be highly effective in the treatment of certain generalized convulsive seizures (65, 66, 67, 68). Among 42 patients with intractable seizures, generalized tonic-clonic seizures were fully controlled in 14 patients by add-on VPA therapy (39). VPA was compared with phenytoin in 61 previously untreated patients with generalized tonic-clonic, clonic, or tonic seizures (69). When seizures occurring before therapeutic plasma drug levels had been reached were discounted, seizures were brought under control in 82% of VPA-treated patients, versus 76% of those treated with phenytoin. In a randomized comparison of VPA and phenytoin in patients with previously untreated tonic-clonic seizures, a 2-year remission was achieved in 27 of 37 patients receiving VPA and in 22 of 39 patients receiving phenytoin (68). Monotherapy with VPA was assessed in two studies of patients with primary (or idiopathic) generalized epilepsies (61,70). Among patients who had generalized tonic-clonic seizures only, complete seizure control was achieved in 51 of 70 patients (70) and in 39 of 44 patients (61), respectively. VPA monotherapy in children with generalized tonic-clonic seizures was also found to be highly effective (71).
Currently, VPA is the agent of first choice for most myoclonic seizures, particularly for those occurring in patients with primary or idiopathic generalized epilepsies (61,62,70). In a group of patients with primary generalized epilepsy given VPA monotherapy, 22 patients had myoclonic seizures and 20 of those experienced at least one other seizure type, either absence or tonic-clonic. The myoclonic seizures were controlled by VPA monotherapy in 18 of the 22 patients (61). Patients with juvenile myoclonic epilepsy have an excellent response to VPA (72), which remains an agent of first choice for this condition. Benign myoclonic epilepsy of infancy, which belongs to the group of primary or idiopathic generalized epilepsies, also responds well to treatment with VPA (71). Some success has been achieved with VPA in patients with postanoxic intention myoclonus (73,74). A combination of VPA and clonazepam is often used to treat the myoclonic and tonic-clonic seizures associated with severe progressive myoclonus epilepsy (75).
Like all other AEDs, VPA is less effective in the treatment of generalized encephalopathic epilepsies of infancy and childhood, such as infantile spasms and Lennox-Gastaut syndrome. In a series of patients treated with VPA, 38 had myoclonic astatic epilepsy—a term used by the authors synonymously with Lennox-Gastaut syndrome (62). Seven patients became and remained seizure free with VPA therapy. A 50% to 80% improvement was achieved in one third of patients, and other AEDs were withdrawn or reduced (62). In the same series, seizures were fully controlled in three of six patients with myoclonic absence epilepsy, all of whom were receiving combination therapy.
Reports on the use of VPA for the treatment of infantile spasms include small numbers of patients (76, 77, 78), or patients receiving corticotropin and VPA simultaneously. With VPA used as the first agent, eight of 19 infants experienced good seizure control and did not require corticotropin treatment (79). The patients who experienced an initial failure with VPA or corticotropin were switched to the other agent. Comparison of the two groups revealed a
tendency toward a better response with corticotropin, but the incidence and severity of side effects was lower with VPA. A low VPA dose of 20 mg/kg per day was used in a series of 18 infants with infantile spasms not previously treated with corticotropin (80). In 12 of these patients, the short-term results were described as good to excellent. On follow-up, seven patients had residual seizure activity. The authors concluded that the efficacy of VPA was similar to that of corticotropin and that VPA was associated with fewer side effects.
tendency toward a better response with corticotropin, but the incidence and severity of side effects was lower with VPA. A low VPA dose of 20 mg/kg per day was used in a series of 18 infants with infantile spasms not previously treated with corticotropin (80). In 12 of these patients, the short-term results were described as good to excellent. On follow-up, seven patients had residual seizure activity. The authors concluded that the efficacy of VPA was similar to that of corticotropin and that VPA was associated with fewer side effects.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


