Figure 50.1. Structure of the 1,4-benzodiazepines. Substituents at the named sites are given in the table for diazepam, lorazepam, clonazepam, nitrazepam, and clorazepate. Midazolam with its fused R1 ring is shown separately. Clobazam is a 1,5 benzodiazepine.
BZ potency correlates with binding affinity at the BZ site on neuronal GABAA receptors (Table 50.1) (14,15). An electron-withdrawing group at the 7 position increases receptor-binding affinity (Table 16) and potency, and all useful anticonvulsant BZs have this feature. A methyl group on the position-1 nitrogen (as in diazepam and clobazam) increases binding affinity and potency, as does a halogen at the 2′ position on the aryl group. A hydroxyl group at position 3 (as in lorazepam) decreases potency and binding affinity.
Table 50.1 Antiseizure Activity, Motor Impairment, and Receptor Binding of Some BZs
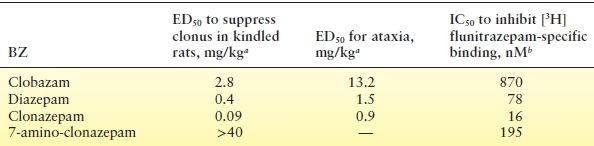
aDose required to inhibit forelimb clonus or to cause ataxia in 50% of amygdala-kindled rats (data from Tietz EI, Rosenberg HC, Chiu TH. A comparison of the anticonvulsant effects of 1,4- and 1,5-benzodiazepines in the amygdala-kindled rat and their effects on motor function. Epilepsy Res. 1989;3:31–40.; (13), except 7-amino-clonazepam data (HC Rosenberg, EI Tietz, and TH Chiu; unpublished data, 1987).
bConcentration required to displace 50% of 2 nM [3H]flunitrazepam specifically bound to rat cerebral cortical membranes (EI Tietz, TH Chiu, and HC Rosenberg; unpublished data, 1990).
BZ activity at the GABAA receptor is a function of the drug’s affinity for the BZ binding site and its intrinsic allosteric effect on the GABAA receptor. The efficacy of individual compounds varies widely. Most BZ ASDs are full agonists that maximally enhance GABAA receptor activity. Competitive antagonists bind to the BZ site but do not affect GABAA receptor function. The BZ antagonist, flumazenil, is used to reverse sedation induced by BZs in anesthesia (17,18) and to treat BZ overdose (19). Several “partial agonists” at the BZ binding site have been characterized, including abecarnil (20), imidazenil (21), and bretazenil (22). Although less effective than full agonists like diazepam, these drugs have demonstrated anticonvulsant efficacy in animal models and appear less prone to the development of tolerance (23,24). Still other compounds, notably the beta-carbolines, behave as “inverse agonists” at the BZ site and inhibit GABA binding or GABA-evoked currents (25). These agents can induce convulsive seizures or anxiety (25,26) and have no clinical utility.
Anticonvulsant Activity
BZs are effective against most experimental seizure types, but there are large differences between individual drugs in their potency, efficacy, and other clinical effects (27). BZs are particularly effective against seizures induced by pentylenetetrazol (28) but are less effective against tonic seizures in the maximal electroshock model (29). BZs also slow the development of kindling (30). The dose ratio between clinical efficacy and adverse effects varies between specific agents. For example, the diazepam dose for blocking pentylenetetrazol seizures is 1% of that necessary to abolish the righting response; for clonazepam, the ratio is <0.02%, suggesting a wider therapeutic window.
BZ Actions at the GABAA Receptor
In 1977, a high-affinity, saturable binding site for BZs was discovered on CNS neuronal membranes (8,9,14). BZ receptor binding was “coupled” to GABA binding (31), which led to the idea of a “GABAA receptor complex” incorporating binding sites for GABA, the BZs, and barbiturates and a ligand-gated chloride channel. Later, it became clear that BZs and GABA bind to sites on a single pentameric channel. Electrophysiologic studies demonstrated that BZs increased the amplitude of GABA-mediated inhibitory postsynaptic potentials (IPSPs) (10) by increasing the opening frequency of the GABA-gated chloride channel (32); this was later confirmed with single-channel studies (33).
In whole-cell patch clamp recordings of isolated CNS neurons, BZs alone produce no GABAA receptor current. Increasing BZ concentrations enhance GABA currents evoked by a low GABA concentration (Fig. 50.2A, C). This enhancement results from an effect of BZs to increase the affinity of GABA at the GABA binding site, which produces a leftward shift of the concentration–response curve for GABA (34) (Fig. 50.2B). There is no change in the kinetics of channel gating (33). The BZs increase the current produced by low GABA concentrations but not by high (millimolar) synaptic concentrations at which receptor binding is saturated (35). Thus, BZs do not usually increase the amplitude of miniature inhibitory postsynaptic currents (mIPSCs) from individual synapses. So, how do they increase GABA currents? By increasing the binding affinity of GABA for the receptor (see Fig. 50.2B), BZs slow the dissociation of GABA from the receptor, which prolongs the mIPSC decay phase (36,37). Prolongation of the mIPSC increases the likelihood of temporal and spatial summation of multiple synaptic inputs, which in turn increases the amplitude of stimulus-evoked polysynaptic IPSCs (Fig. 50.2D). The BZs thus increase the inhibitory “tone” of GABA-ergic synapses, which reduces the hypersynchronous firing of neuron populations that underlies seizures (38).
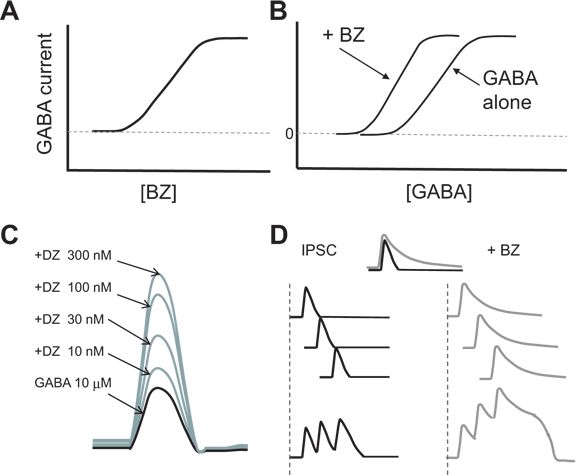
Figure 50.2. Effects of BZs on GABAA receptor currents. A: Increasing BZ concentration with a single low concentration of GABA evokes progressively larger currents, as shown in (C) for increasing diazepam (DZ) concentrations (mock data). B: Addition of a single BZ concentration to increasing GABA concentrations shifts the concentration–response curve for GABA to the left, indicating increased affinity for GABA at the receptor but no increase in maximal current. The BZs have no effect in the absence of GABA. D: The increased GABA affinity prolongs the IPSC decay phase (gray), resulting in increased temporal and spatial summation to produce larger polysynaptic currents (mock data).
Molecular Biology of GABAA Receptors
GABAA receptors are pharmacologically complex, with binding sites for BZs; barbiturates; neurosteroids; general anesthetics; the novel anticonvulsant, loreclezole; and the convulsant toxins, picrotoxin and bicuculline. Protein subunits from seven different subunit families (39) assemble to form pentameric (40) transmembrane chloride channels (Fig. 50.3). In mammals, 16 subunit subtypes have been cloned, including 6 α, 3 β, and 3 γ subtypes, as well as δ, π , ε, and θ, and there are alternatively spliced variants of the β2 and γ2 subtypes (39). Though thousands of subunit compositions are possible, expression is regulated by region and cell type (41) and also developmentally controlled (42), reducing the number of possible isoforms in specific brain regions and individual neurons. The most common GABAA receptor has a presumed stoichiometry of two α1, two β2, and a single γ2 subunit; the δ subunit may in some cases substitute for γ, particularly when receptors are expressed extrasynaptically. The subunits are arranged around a central water-filled pore, which opens to conduct Cl− ions when GABA is bound (see Fig. 50.3). Studies of recombinant receptors have shown that individual subunit subtypes confer different sensitivities to GABAA receptor modulators, including BZs (43), loreclezole (44), and zinc ions (45).
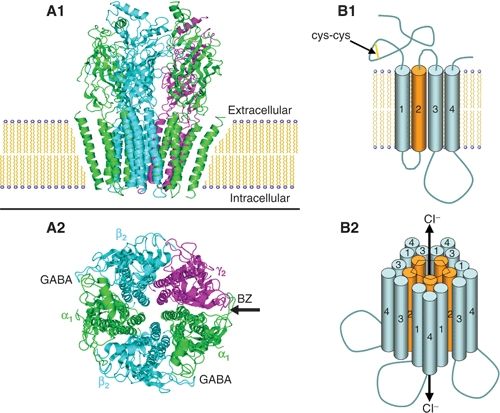
Figure 50.3. Model of a GABAA receptor in the plasma membrane. A: A space-filling model of the pentamer in side view (A1) and top view (A2) based on the high sequence homology with the nicotinic acetylcholine receptor. There are with two binding sites for GABA, between α and β subunits, and one for BZs, between the alpha and gamma subunits (arrow). B: A schematic view shows the topology of each subunit with a large extracellular loop containing a cysteine loop (B1) and four transmembrane domains from which the second forms the lining of the chloride ion channel. Binding of GABA allows the channel to open and conduct Cl− ions (B2), resulting in the fast inhibitory postsynaptic potential (IPSP)
GABAA Receptor Subunits and BZ Pharmacology
BZ augmentation of GABAA receptor currents requires a γ subunit, and the selectivity of BZ responsiveness is determined by which α subunits are present (46). The BZ binding site is located in a cleft between the extracellular amino termini of these two subunits. The α1 subunit results in a receptor with high affinity for the hypnotic, zolpidem, defining the “BZ-1” (or Ω1) receptor type (47). The α2 and α3 subunits result in receptors with moderate zolpidem affinity, termed BZ-2 receptors. GABAA receptors with the α5 subunit and/or the γ3 subunit are sensitive to diazepam, but far less so to zolpidem, and are termed BZ-3 receptors. GABAA receptors with the α4 or α6 subunits are insensitive to most BZs (46).
The GABAA receptor subunit composition determines not only the affinity for particular BZs but also the clinical/behavioral effects of the BZ at that specific receptor subtype. The role of the α subunits in BZ pharmacology was revealed by the discovery of a single histidine (H) residue found in all BZ-sensitive α subunits (H101 in the rat α1 subunit) but not in the BZ-insensitive α4 or α6 subunits, which instead have a charged arginine (R) residue. This H residue was discovered in a strain of “alcohol-nontolerant” rats, which were found to have a spontaneous point mutation in the α6 subunit (R100Q) that made their α6-containing GABAARs (found mostly in the cerebellum) diazepam sensitive. Since alcohol appears to enhance GABAAR function via the BZ site (39), the abnormally BZ-responsive receptors in the cerebellum resulted in ataxia and intolerance to these agents (48). Mutation of R100 to H in α6 dramatically increased BZ binding in this normally insensitive subunit, while mutation of H101 to R in α1 reduced BZ sensitivity (49). BZ-insensitive α subunit mutations were subsequently “knocked in” to identify BZ actions at receptors containing that subunit. In homozygous α1(H101R) knock-in mice, the anxiolytic effect was intact, but BZs were not protective against pentylenetetrazol-induced convulsions and did not produce sedation or amnesia, suggesting that binding to the (wild type) α1 subunit is responsible for sedative, amnestic, and antiseizure actions (50). Moreover, the α1-selective sedative–hypnotic, zolpidem, showed no sedative effect in α1(H101R) mice (51). Unfortunately, these findings underscore the association between sedation and antiseizure efficacy at α1-containing GABAARs. Similarly, the anxiolytic (52) and myorelaxant (53) properties of BZs appear to derive from α2- and α3-containing GABAARs, while the α5 subunit was critical for amnestic effects (54). BZs may also have a true analgesic effect independent of their sedative and anxiolytic actions, associated with the α2 and α3 more than α5 subunits (55). Since there is no evidence of biophysically distinct effects of BZs on receptors composed of different α subunits, the different behavioral effects are likely due to the brain regions and neuronal populations expressing specific GABAAR isoforms. New α2/α3 subunit–selective BZs appear to have anxiolytic activity without causing sedation (56). Development of nonsedating antiseizure BZs that do not induce tolerance (57) may also be possible.
GABAA Receptors and Epilepsy
The anticonvulsant properties of BZs are likely related to the prominent role of GABAA receptors in epilepsy. The evidence linking epilepsy with dysfunction of GABA-ergic inhibition is substantial (38). GABAA receptors are the target not only of the BZs but of other ASDs including the barbiturates and, indirectly, of two agents that increase GABA concentration at the synapse, tiagabine and vigabatrin (38). Several animal models of epilepsy have demonstrated altered GABAA receptor number or function (38,58). Moreover, genetic or acquired changes in the composition or structure of the transmembrane protein subunits that make up GABAA receptors can result in epilepsy. GABAA receptor subunit expression is altered in the hippocampi of experimental animals with recurrent seizures (59) and in patients with temporal lobe epilepsy (60,61). Reduced expression of the GABAAR γ2 subunit in rats using an antisense oligonucleotide to block translation of endogenous γ2 mRNA led to spontaneous electrographic seizures that evolved into limbic status epilepticus (62). In humans, Angelman syndrome, a neurodevelopmental disorder associated with severe mental retardation and epilepsy, is linked to a deletion mutation on chromosome 15q11-13 (63) in a region encoding the GABAA receptor β3 subunit (64). In addition, two mutations in the γ2 subunit that impair GABAA receptor function (65), K289M (66) and R43Q (67), have been linked to a human syndrome of childhood absence epilepsy and febrile seizures, and a loss-of-function mutation in the α1 subunit was found in a family with autosomal dominant juvenile myoclonic epilepsy (68). The R43Q mutation in the γ2 subunit reduces BZ sensitivity (69) by altering GABAA receptor assembly (70) and trapping the receptor in the endoplasmic reticulum (71). The relative positions of these and several other epilepsy-causing mutations are shown schematically on a “generic” GABAA receptor subunit in Fig. 50.4, along with the sites of action for several GABAA receptor subunit–associated ASDs (72).
Other BZ Actions
With a few caveats (26), the BZs derive their anticonvulsant properties from their specific interaction with GABAA receptors. At doses used to treat status epilepticus, BZs can also inhibit voltage-gated sodium (73) and calcium channels (74) and can increase GABA levels in cerebrospinal fluid (75). However, it should be noted that the BZs have no interaction with the G protein–linked GABAB receptor, which can either suppress voltage-gated Ca2+ channels or activate inward rectifying K+ channels (76).
The BZs also bind to the “peripheral BZ receptor” (PBR) (77), an 18-kDa protein in the outer mitochondrial membrane that functions as part of the mitochondrial permeability transition pore involved in cholesterol transport (77), apoptosis, and regulation of mitochondrial function (78). Although the PBR is widely expressed throughout the body, its expression in the CNS is restricted to ependymal cells and glia (79); hence, it is unlikely that the PBR is involved in the antiseizure properties of the BZs, though it may explain the neuroprotective properties of some BZs (80,81).
Excitatory GABAA Currents
BZ enhancement of GABAA receptor function may not always be anticonvulsant or even inhibitory. Early in CNS development, neurons express the Na+/K+/Cl− cotransporter, NKCC1, rather than the K+/Cl− cotransporter, KCC2, which is expressed in adult neurons. NKCC1 increases intracellular Cl− resulting in a depolarizing Cl− reversal potential, while KCC2 exports Cl− yielding the hyperpolarizing Cl− reversal potential found in adult neurons (82). As a result, activation of GABAA receptor channels can be excitatory during early development (83) and play a trophic role in neuronal migration and connectivity (84,85), but this may also contribute to epileptogenesis (86). In fact, endogenous GABA appears to be proconvulsant in early postnatal rat hippocampal slices, as GABAA antagonists blocked epileptiform activity induced by depolarization with high external [K+] (87). However, BZ anticonvulsant efficacy appears to be intact, likely because persistent opening of GABA channels (in the presence of BZs) may reduce the depolarizing chloride reversal potential, resulting in “shunt” inhibition, or alternatively, subthreshold GABA-evoked depolarization may inactivate sodium channels and prevent action potential firing (88). The current through GABAA receptor channels can also be altered by changes in intracellular bicarbonate, [HCO3−] (82), which, like Cl−, can flow through the channel (89). Changes in [HCO3−] may underlie reduced synaptic GABA currents during development of BZ tolerance (90). Depolarizing GABAA currents may also be a source of interictal spike activity, as observed in epileptic subiculum neurons in brain slices of hippocampi removed from patients with temporal lobe epilepsy (91). Changes in the GABA current reversal potential might also explain why diazepam may be less effective in children with epileptic encephalopathies (92) and rarely can cause status epilepticus in patients with the Lennox–Gastaut syndrome (93).
ABSORPTION, DISTRIBUTION, AND METABOLISM
The major anticonvulsant role of the BZs is in the treatment of status epilepticus (SE) and seizure clusters, for which they represent first-line therapy, preferably administered intravenously (IV) (94). In very young children, this may be difficult or impossible, necessitating administration via rectal (95–100), intraosseous (101), buccal (102,103), or nasal (104–106) routes. With IV administration, the main factor in a drug’s effectiveness for SE is the rate at which it crosses the blood–brain barrier (BBB). The BZs are highly lipophilic and cross the BBB rapidly (107), though this varies more than 50-fold between agents (108) and is fastest for the most lipophilic agents, such as diazepam. Protein binding also correlates with lipophilicity and is high for most BZs, up to nearly 99% for diazepam. The BZs are fully absorbed after oral ingestion.
Despite generally long plasma half-lives, most BZs are relatively “short-acting” after administration of a single dose due to a similarly rapid distribution from the brain and vascular compartments to peripheral tissues (109,110). BZ pharmacokinetics are best fit by a two-compartment model: high levels occur rapidly in the brain and other well-perfused organs and then decline rapidly with an initial brief half-life due to distribution into peripheral tissues and lipid stores, followed by a much slower elimination half-life related to enzymatic metabolism and excretion. For example, the elimination half-life of diazepam ranges from 20 to 54 hours (111), but the duration of action after a single IV injection is only 1 hour, with peak brain concentrations present for only 20 to 30 minutes (112).
The BZs are metabolized in the liver by cytochrome P450 enzymes, particularly CYP3A4 and CYP2C19, with relatively little enzyme induction. The presence of biologically active metabolites (e.g., nordazepam, a metabolite of diazepam) can significantly prolong the biologic half-lives of some BZs. Elimination may be prolonged by enterohepatic circulation, particularly in the elderly. Most BZs cross the placenta and are secreted into breast milk. A schematic of BZ metabolic pathways is shown in Figure 50.5. The biotransformation and pharmacokinetics of the BZs have been extensively reviewed (115–117) and will be presented in more detail for the individual agents below.
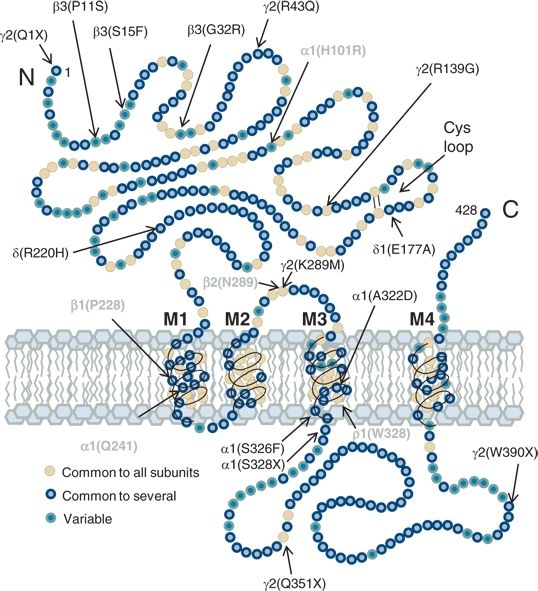
Figure 50.4. Model of a prototype GABAAR subunit (based on α1 subunit diagram from Olsen RW, Tobin AJ. Molecular biology of GABAA receptors. FASEB J. 1990;4:1469–1480.) showing approximate locations of point mutations associated with generalized epilepsies (see also Macdonald RL, Kang JQ, Gallagher MJ. GABAA receptor subunit mutations and genetic epilepsies. In: Noebels JL, Avoli MB, Rogawski MA, Olsen RW, Delgado-Escueta AV, editors. Jasper’s Basic Mechanisms of the Epilepsies [Internet]. 4th edition. Bethesda (MD): 2012) in black and point mutations associated with ASD sites of action (see text for details).
DRUG INTERACTIONS
BZs interact with other drugs more prominently through pharmacodynamic than pharmacokinetic mechanisms. They do not significantly affect plasma protein binding or metabolism of other drugs. CNS depression is increased when BZs are given in conjunction with other CNS-depressant drugs (118). Pharmacokinetic interactions with other anticonvulsants are infrequent and inconsistent, with the exception of phenobarbital. Diazepam enhances phenobarbital elimination (119), and phenobarbital increases clearance (120) and lowers plasma levels of clonazepam (121). Clobazam increases the 10,11-epoxide metabolite of carbamazepine (122). Valproate reduces diazepam protein binding, increasing free drug levels (123), and enhances diazepam’s CNS effects (119). Other ASDs may augment metabolism and clearance of N-desmethyldiazepam derived from clorazepate (124). Inhibitors of CYP3A4, including erythromycin, clarithromycin, ritonavir, itraconazole, ketoconazole, nefazodone, and grapefruit juice, can slow BZ metabolism (125). Cimetidine decreases the clearance of diazepam (126,127) and nitrazepam (128). Rifampin increases the clearance and shortens the half-life of nitrazepam (129). Lorazepam half-life is markedly increased by probenecid (130).
ANTISEIZURE EFFICACY
Status Epilepticus
Status epilepticus is associated with significant morbidity and mortality (131,132) and requires emergent medical treatment to avoid neuronal damage and its neurologic consequences (133,134). The BZs have become agents of choice for initial therapy due to their rapid onset, proven efficacy (135), and lower risk of cardiotoxicity or respiratory depression compared to the barbiturates (136). The role of BZs in status epilepticus has been confirmed in several well-controlled clinical trials, including the multicenter, double-blind VA Cooperative Status Epilepticus Trial (137).
Lorazepam and diazepam were compared for treatment of status epilepticus in a double-blind study of 78 adults with epilepsy (135). Intravenous lorazepam (4 mg) stopped status epilepticus in 78% of patients and diazepam (10 mg) in 58% after the first injection; both had similar efficacy (89% and 76%, respectively) after the second injection. An open-label, prospective, randomized trial compared lorazepam (0.05 to 0.1 mg/kg) and diazepam (0.3 to 0.4 mg/kg) in children with acute convulsions, including convulsive status epilepticus (138). Lorazepam was more effective (P < 0.01) after the first dose and apparently safer than diazepam. A meta-analysis of 11 randomized controlled trials with 2017 participants found that lorazepam was better than either diazepam or phenytoin alone for reducing risk of seizure continuation (139). A population-based study of 182 children with convulsive status epilepticus showed that IV lorazepam was 3.7 times more likely than rectal diazepam to terminate seizures (140). Lorazepam’s superiority may be due to its longer duration of action, based on a longer distribution half-life (see Lorazepam: Pharmacokinetics).
Lorazepam has largely replaced diazepam as the agent of choice for prehospital treatment of status epilepticus. A prehospital trial of lorazepam and diazepam found that status epilepticus had terminated by arrival at the emergency department in 59.1% of patients treated with lorazepam (2 mg), compared to 42.6% of patients treated with diazepam (5 mg) and 21.1% of patients given placebo (141). Rates of circulatory or ventilatory complications for lorazepam and diazepam were similar (10.6% and 10.3%, respectively) and lower than that of placebo (22.5%), confirming the safety of BZ treatment in this setting. However, patients treated with >30% more than the standard therapeutic BZ doses were more likely to require intubation for airway protection and had significantly longer hospitalizations (2 weeks vs. 1 week) (142).
Early treatment of status epilepticus increases the probability of seizure termination (141), likely because prolonged seizures lower GABAA receptor sensitivity to BZs (143). Reduction in GABAA receptor BZ sensitivity can occur within minutes in status epilepticus (144,145) and may be responsible in part for both the persistent epileptic state and its refractoriness to treatment. Refractoriness to BZs may be mediated by N-methyl-d-aspartate (NMDA) receptor mechanisms, as NMDA antagonists improve the response to diazepam in late pilocarpine-induced status epilepticus (146). These findings suggest a possible strategy for treatment of late, BZ-refractory status epilepticus with combinations of a BZ and an NMDA receptor antagonist such as the dissociative general anesthetic, ketamine. Efficacy of combined diazepam and ketamine has been demonstrated in a rat model of status epilepticus (147). A recent nonrandomized trial of oral ketamine for refractory nonconvulsive status epilepticus in children showed efficacy in five of five cases (148). Treatment protocols involving NMDA antagonists have been suggested (149,150), but such approaches will require validation in controlled clinical trials.
Both lorazepam and diazepam have been approved by the U.S. Food and Drug Administration (USFDA) for treatment of status epilepticus in adults; diazepam has also been approved in children older than 30 days. Parenteral preparations of other BZs, including midazolam, flunitrazepam, and clonazepam, expand the possibilities for BZ treatment of status epilepticus. However, parenteral clonazepam is currently available only in Germany and the UK, and flunitrazepam is not available in the USA. Alternative routes of administration, including intramuscular (IM) injection and intranasal (104,105), buccal (102), endotracheal (151,152), or rectal (100,153,154) instillation, also rapidly produce therapeutic levels and have demonstrated efficacy against status epilepticus or seizure clusters.
Acute Repetitive Seizures
The availability of alternative methods of BZ administration increases the therapeutic options for treatment of acute repetitive seizures. Individual agents can be selected for specific clinical situations. For example, repeated seizures in a patient rapidly tapered off anticonvulsants for inpatient epilepsy monitoring could be treated with diazepam (rather than lorazepam) since its shorter peak duration of action may be less likely to suppress seizure activity needed later for localization of seizure onset. In the case of serial seizures, the need for high drug levels immediately is less urgent, and the ease of administration by family or allied health workers becomes important. Diazepam rectal gel is effective in preventing subsequent seizures during seizure clusters (153–155) and can reduce the frequency of emergency department visits (96). Buccal (156,157) and intranasal (158) routes may be equally effective and more acceptable (159). Table 50.2 compares the clinical and pharmacologic properties of the BZs used for acute seizures.
Table 50.2 Clinical Pharmacology of BZs Used for Acute Seizures
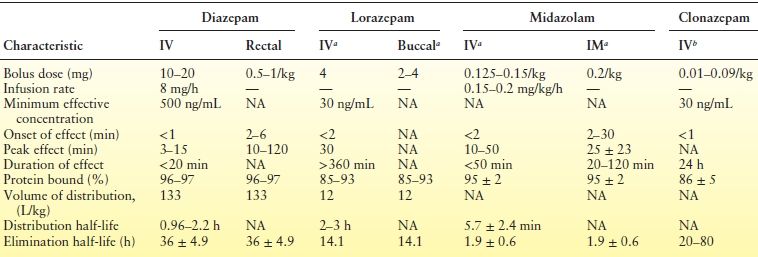
aNot approved by the USFDA for seizures.
bNot available in the United States.
NA, not available.
Chronic Treatment of Epilepsy
Although the use of BZs in chronic treatment of epilepsy is limited by sedation and the development of tolerance, BZs may have specific therapeutic indications, such as adjunctive treatment of myoclonic and other generalized seizure types or in conjunction with comorbid anxiety disorders. For example, lorazepam improved control of seizures associated with psychological stressors (160). Intermittent use of BZs when seizure thresholds are transiently reduced may be the ideal strategy for these ASDs. Not only are they suited pharmacokinetically for such applications, but short-term use may avoid the development of tolerance. For example, catamenial seizures improved with intermittent administration of clobazam (161). ASD efficacy for specific indications will be discussed with the individual agents below.
TOLERANCE AND DEPENDENCE
BZ Tolerance
Chronic BZ treatment is associated with tolerance, a decrease in sedative or anticonvulsant properties, and dependence, the need for continued drug to prevent a withdrawal syndrome (162). The development of tolerance is a significant clinical problem, requiring escalation of drug doses and increasing the risk of withdrawal seizures. Chronic treatment with BZs can also reduce their subsequent effectiveness in acute conditions (163), rendering them less useful for treatment of status epilepticus. In animal studies, tolerance develops proportionally to agonist efficacy. BZ partial agonists develop much less tolerance than full agonists, and the antagonist flumazenil causes no tolerance-related changes in receptor number or function (24). Tolerance to one BZ with a particular regimen may not induce tolerance to a different BZ, suggesting drug-specific interactions of each agent at BZ receptor sites on specific GABAA receptor subunit combinations (164). The duration of tolerance also varies between BZs (165). Several studies of tolerance have noted changes in GABAA receptor subunit expression (166–168) as well as functional changes (169,170); however, such changes are dependent not only on the drug and dosage but also on the duration and method of drug administration, all of which contribute to chronic BZ receptor occupancy that predisposes to tolerance. Measurements of tolerance also depend on the animal seizure model and the behavioral tests used to assess BZ clinical properties (171).
Physical Dependence
Abrupt cessation of prolonged BZ therapy can result in withdrawal symptoms, including restlessness and agitation, anxiety, loss of appetite, nausea, lethargy, dizziness, headache, palpitations, irritability, confusion, and, in some cases, seizures. The short-acting antagonist, flumazenil, precipitated a withdrawal syndrome in subjects given chronic low-dose diazepam (mean dose 11.2 mg/day) for an average of 4.6 years (172). Four of 13 patients developed panic attacks. A short-lived withdrawal syndrome was elicited by IV flumazenil following 7, 14, or 28 days of oral diazepam (15 mg/70 kg) administration to healthy volunteers (173). There is debate whether withdrawal symptoms, such as heightened anxiety, might represent rebound of existing symptoms to a level greater than that before treatment and whether withdrawal anxiety can result in relapse to the previous state of anxiety (174). BZ prescription misuse has been ascribed to patients’ efforts to alleviate withdrawal symptoms, which can lead to a drug dependence syndrome (175,176). BZ self-administration is enhanced in long-term therapeutic users suddenly switched to placebo relative to those whose dose was tapered gradually (177).
Changes in GABAA receptors related to tolerance might be assumed to underlie withdrawal symptoms and physical dependence (178,179). Yet, evidence from animal models suggests that enhancement of excitatory systems in a variety of brain regions (180) may underlie anxiety behavior (181–183) and increased seizure activity (183). Enhanced glutamatergic neurotransmission involves a selective increase in GluR1-containing alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA) receptors (183–185), which correlates with increased anxiety-like behavior (181). Such neuroplastic changes in the hippocampus are similar to those found with long-term potentiation (LTP) (186). LTP depends on AMPA receptor–mediated depolarization and subsequent relief of the Mg2+ block from NMDA receptors, allowing Ca2+ entry, which in turn activates kinases resulting in persistent enhancement of GluA1 AMPA receptors. In contrast, for BZ withdrawal, the calcium signal that mediates enhancement of GluA1 AMPA receptors enters through voltage-gated calcium channels (VGCCs) (181,182). In fact, VGCC currents double during chronic BZ treatment and withdrawal, and anxiety can be alleviated by prior administration of the VGCC antagonist, nimodipine (182). The increased anxiety and other symptoms abate over time, associated with downregulation of NMDA receptors (181,187,188), which may serve as a natural brake on withdrawal symptoms.
ADVERSE EVENTS
With acute treatment of status epilepticus, the primary adverse effects are respiratory and cardiovascular depression (135,189). For IV infusions, the propylene glycol solvent contributes to toxicity (190). Other toxic effects such as sedation and amnesia are of relatively little consequence in this setting and difficult to distinguish from the effects of status epilepticus itself. However, sedation after termination of convulsive status epilepticus often necessitates EEG evaluation to ensure that convulsive seizures have not been converted to nonconvulsive status epilepticus. When BZs have been administered in conjunction with other CNS-active drugs such as phenobarbital, respiratory and cardiovascular toxicity may be enhanced (118). In patients with Lennox–Gastaut syndrome, parenterally administered BZs can induce tonic status epilepticus, though this is rare (191). Thrombophlebitis may occur (192), and intra-arterial injection may produce tissue necrosis (193).
With chronic use, all of the BZs induce similar untoward effects including sedation and drowsiness, lightheadedness, ataxia, cognitive slowing and confusion, and anterograde amnesia. Other effects include weakness, headache, blurred vision, vertigo, nausea and vomiting, GI distress, and diarrhea. Joint aches, chest pains, and incontinence occur more rarely (108). The risk of tolerance, dependence, and abuse is significant but low in patients prescribed these agents for appropriate indications (110,194). Abrupt withdrawal of the BZs has been associated with convulsions, worsening of insomnia, psychosis, and delirium tremens in nonepileptic individuals using diazepam, clonazepam, clorazepate, or nitrazepam (195,196). The incidence of allergic, hepatotoxic, or hematologic reactions to the BZs is extremely low. The BZs can sometimes increase the frequency of seizures in epileptic patients. Specific adverse effects will be discussed with the individual agents below.
INDIVIDUAL BZS
Diazepam, lorazepam, and midazolam are used predominantly for short-term indications including status epilepticus, serial seizures, and episodes of lower seizure threshold. Clonazepam, clorazepate, clobazam, and nitrazepam are used more commonly for treatment of chronic epilepsy.
Diazepam
Diazepam, the first BZ used in the treatment of epilepsy (5), became a standard initial therapy for status epilepticus in adults and children (135), though its primary role has been usurped by lorazepam (137,141). Diazepam is available in both oral and parenteral preparations. The classification of rectal diazepam as an orphan drug in 1993 allowed the development of rectal diazepam gel (Diastat).
Diazepam and other BZs induce an increase in β-frequency activity and slowing of the background on EEG, which can be quantified by spectral analysis (197). The pattern of EEG changes may be of prognostic value in seizure control; 88% of patients (29/33) whose EEG responded to diazepam with loss of abnormal activity or emergence of fast (β-frequency) activity had a good prognosis (seizure free or 50% seizure reduction) (198).
Absorption, Distribution, and Metabolism
Diazepam is highly lipophilic, which allows rapid entry into the brain but also results in rapid subsequent redistribution into peripheral tissues. It is extensively bound to plasma proteins (90% to 99%) (199). The volume of distribution is 1.1 L/kg. Plasma concentration declines rapidly during the distribution phase with an initial half-life (t1/2α) of 1 hour (200). Diazepam undergoes demethylation to desmethyldiazepam (DMD, nordazepam), a metabolite with anticonvulsant activity and a long half-life (>20 hours), followed by slow hydroxylation to oxazepam, which is also active (see Fig. 50.5) (201). Small amounts of temazepam are also formed by 3-hydroxylation of diazepam. The hydroxylated metabolites are conjugated with glucuronic acid in the liver (202) followed by renal excretion (201) with an elimination half-life (t1/2β) of 24 to 48 hours (119,200). Diazepam treatment causes modest induction of cytochrome P450 type 2B (203). There is little evidence of enterohepatic circulation (204), but diazepam may be secreted in the gastric juices resulting in enterogastric circulation (205). Like most BZs, diazepam crosses the placenta and is excreted in breast milk (108,200).
Adverse Effects and Drug Interactions
Diazepam can produce respiratory depression (206), which may be exacerbated by postictal CNS depression and necessitate ventilatory support (207). Sedative effects, including inattention and drowsiness, occur at plasma levels of about 200 ng/mL (208), the same level needed to suppress spikes (209) and maintain control of status epilepticus in acute studies (119). Drowsiness, fatigue, amnesia, ataxia, and falls are more prominent in the elderly. Intravenous diazepam can cause thrombophlebitis and lactic acidosis (due to the propylene glycol vehicle) (192,210). Rare paradoxical responses include increased seizure frequency, muscle spasms, or status epilepticus (125). An idiosyncratic allergic interstitial nephritis has also been reported (211). Other rare adverse events include cardiac arrhythmias, hepatotoxicity, gynecomastia, blurred vision and diplopia, neutropenia or thrombocytopenia, rash and urticaria, and anaphylaxis (212). There is potential for abuse, though it is rare in patients prescribed diazepam for appropriate indications (110). The teratogenicity of diazepam is uncertain, but diazepam taken during the first trimester has been associated with oral clefts (213). Diazepam may also amplify the teratogenic potential of valproic acid (214).
Diazepam enhances the elimination of phenobarbital (119), likely due to induction of cytochrome P450 (203). Valproic acid displaces diazepam bound to plasma proteins, leading to increased free diazepam and associated increased sedation (123).
Clinical Applications
Status Epilepticus.
Diazepam is effective initial therapy in both convulsive and nonconvulsive status epilepticus (137). It may be particularly effective in generalized absence status epilepticus, with 93% of patients initially controlled (215). In the same early study, diazepam also controlled 89% of generalized convulsions, 88% of simple motor seizures, and 75% of complex partial status epilepticus. These numbers are higher than those observed in the VA Cooperative Status Epilepticus Trial (137), possibly due to differences in patient populations.
Diazepam is typically administered initially as a single IV bolus of 10 to 20 mg (119). A 20-mg bolus given at a rate of 2 mg/minute stopped convulsions in 33% of patients within 3 minutes and in 80% within 5 minutes (216), but a single injection often does not produce lasting control, due to its short duration of action, and may be less effective when status results from acute CNS disease or structural brain lesions (115). Strategies to avoid this problem have included giving subsequent 5 to 10 mg IV doses every few hours, following diazepam with a longer-lasting anticonvulsant (e.g., phenytoin (137)), or continuous IV diazepam. Repeated dosing results in a decrease in apparent volume of distribution and clearance; hence, subsequent doses should be tapered to prevent toxicity (217). Diazepam (100 mg in 500 mL of 5% dextrose in water) infused at 40 mL/hour delivers 20 mg/hour (94) and may be suitable to obtain a serum level in the range of 200 to 800 ng/mL; 500 ng/mL appears to be effective for termination of status epilepticus (119,218). Complete suppression of 3 Hz spike and wave required 600 to 2000 ng/mL (219). Continuous infusion has been used in patients hypersensitive to anticonvulsants (220). Diazepam is absorbed onto polyvinyl chloride bags, with a reduction in bioavailability of 50% after 8 hours (221), which should be taken into account if a chronic infusion of diazepam for status epilepticus is contemplated. As noted above, the efficacy of BZs decreases with duration of status epilepticus (143); hence, higher levels or alternative treatments may be necessary if seizures are refractory.
Pediatric Status Epilepticus.
The initial recommended IV diazepam dose in children is 0.1 to 0.3 mg/kg IV by slow bolus (<5 mg/minute) repeating every 15 minutes for two doses, with a maximum of 5 mg in infants and 15 mg in older children (222). If IV administration is not possible, a diazepam solution (0.5 to 1 mg/kg), placed 3 to 6 cm into the rectum, has been effective (97). Continuous IV diazepam infusion (0.01 to 0.03 mg/kg/min) controlled seizures in 86% of pediatric patients in status epilepticus (49/57) within an average of 40 minutes (223). Hypotension occurred in 1 patient (2%), respiratory depression in 6 patients (12%), and death in 7 patients (14%). A meta-analysis of 111 pediatric patients (1 month to 18 years) with refractory generalized convulsive status epilepticus, treated with diazepam, midazolam, thiopental, pentobarbital, or isoflurane, suggested that diazepam was less effective as continuous therapy than the other agents (86% vs. 100%) after stratifying for etiology of status epilepticus (224). However, all of the patients receiving diazepam were from one region (India), and none received continuous EEG monitoring, suggesting that differences in location or details of care may have been contributory. Mortality was 20% in symptomatic cases and 4% in idiopathic cases and was less frequent in midazolam-treated patients.
Although IV administration is preferable, rectal administration of diazepam rapidly produces effective drug levels (225) and safely aborts status epilepticus in pediatric patients (95,99). In children found to be in electrographic status epilepticus during EEG monitoring, rectal diazepam stopped paroxysmal activity in 58% of cases (226). Rectal diazepam was particularly effective in patients with electrical status epilepticus during sleep and less effective in patients with hypsarrhythmia. Intraosseous injection is a viable alternative in children of suitable age when IV access is not available (101).
Febrile Convulsions.
Rectal diazepam is effective in aborting febrile and nonfebrile seizures in the home setting (96). While the concept of chronic prophylaxis for childhood febrile convulsions has long been in disrepute, it has not been clear whether there was benefit to short-term seizure prophylaxis during fever. A prospective trial randomized 289 children in Denmark to intermittent prophylaxis (diazepam at fever) or no prophylaxis (diazepam at seizure) and assessed neurologic outcome; motor, cognitive, and scholastic achievement; and likelihood of future seizures 12 years later (227). There were no differences in these measures, suggesting that short-term prophylaxis is probably not necessary. Moreover, the incidence of respiratory depression in children treated with IV and/or rectal diazepam is fairly high, with 9% showing decreased respiratory rate or oxygen saturation in one study (207).
Acute Repetitive Seizures.
In a large-scale multicenter open- label trial of rectal diazepam gel (Diastat) in 149 patients older than 2 years, 77% of diazepam administrations resulted in no further seizures for the ensuing 12 hours (154). There was no loss of effectiveness with more frequent (>8) doses, suggesting that tolerance did not reduce the effectiveness of diazepam under these conditions. Sedation occurred in 17%. Diazepam rectal gel was also useful against serial seizures in adult patients with refractory epilepsy (100,153), with 0.5 mg/kg found to be an effective dose (98). Intramuscular diazepam injection may also be suitable for prophylaxis of serial seizures, but absorption is not rapid enough to be effective against status epilepticus. Intranasal diazepam administration is another alternative in this setting. In healthy human volunteers, peak serum concentrations of diazepam (2 mg) after intranasal administration occurred after 18 ± 11 minutes with bioavailability of about 50% (104). A pharmacodynamic effect was seen at 5 minutes.
Chronic Epilepsies.
Periodic courses of diazepam have been proposed as therapy for several chronic conditions, including West syndrome, Lennox–Gastaut syndrome, Landau–Kleffner syndrome, and electrical status epilepticus during sleep (228). Oral diazepam (0.5 to 0.75 mg/kg/d) administered in cycles of 3 weeks duration was beneficial in interrupting electrical status epilepticus and improved neuropsychological function in some cases.
Lorazepam
Lorazepam (see Fig. 50.1) has greater potency and a longer duration of action than diazepam and has become the agent of choice for initial treatment of status epilepticus in adults (132). Lorazepam is also less likely to produce significant respiratory depression (138). It is available in both oral and parenteral preparations.
Pharmacokinetics
Lorazepam is rapidly absorbed but less bioavailable after oral administration due to first-pass biotransformation in the liver (229). Peak plasma levels occur 90 to 120 minutes after oral dosing (230). Lorazepam is about 90% protein bound, with CSF levels approximately equivalent to free serum levels (231). Sleep spindles in EEG recordings were observed within 30 seconds to 4 minutes after IV lorazepam (232), though peak brain concentrations and maximal EEG effect did not occur until 30 minutes (233). The volume of distribution is about 1.8 L/kg (233). After a single IV injection, plasma levels decrease initially due to tissue distribution with a half-life (t1/2α) of 2 to 3 hours. The minimal effective plasma concentration for control of status epilepticus was 30 ng/mL (234); after IV injection of 5 mg, plasma levels remain above that level for about 18 hours (235). Sedation, amnesia, and anxiolysis occur at plasma levels between 10 and 30 ng/mL (230).
Lorazepam is metabolized in the liver via glucuronidation at the 3-hydroxy group (236) and then excreted by the kidneys (237) (see Fig. 50.4). The half-life for elimination (t1/2β) is in the 8 to 25 hours range (mean 15 hours) and is the same for oral administration (238).
Adverse Effects and Drug Interactions
Sedation, dizziness, vertigo, weakness, and unsteadiness are relatively common, with disorientation, depression, headache, sleep disturbances, agitation or restlessness, emotional disturbances, hallucinations, and delirium less common (239). Psychomotor impairment, dysarthria, and anterograde amnesia have also been observed. Lorazepam (0.2 mg orally) impaired driving more than a blood alcohol level of 0.5 mg% (240). Mild respiratory depression sometimes occurs, particularly with the first IV dose (241). Rare adverse events include neutropenia. A paradoxical effect was observed in a patient with Lennox–Gastaut syndrome in which lorazepam precipitated tonic seizures (242). Abuse liability is relatively low. Although lorazepam is in USFDA pregnancy category D, of unknown teratogenic potential, short-term use in treatment of status epilepticus may be of lifesaving benefit and likely to outweigh the uncertain risks. Sudden discontinuation after chronic use has caused withdrawal seizures (243).
Valproic acid increased plasma concentrations of lorazepam (244) and decreased lorazepam clearance by 40% (245), apparently by inhibiting hepatic glucuronidation. Lorazepam does not affect valproic acid levels (244). Probenecid increased the half-life of lorazepam by inhibiting glucuronidation, resulting in toxicity in patients on long-term therapy (130).
Clinical Applications
Status Epilepticus.
The recommended IV dose of lorazepam for status epilepticus is 0.1 mg/kg (up to a maximum of 4 mg) administered at 2 mg/minute, with repeat doses after 10 to 15 minutes if necessary (137). Although lorazepam is less lipophilic than diazepam, it crosses the BBB readily. Onset of action occurred within 3 minutes, with control of status epilepticus in 89% of episodes within 10 minutes (135). In another study, all 10 patients with generalized convulsive status epilepticus had seizures controlled with IV lorazepam (mean 4 mg), but 9 of 11 patients with complex partial status epilepticus experienced problems, including respiratory depression (246). Other studies have shown response rates of 80% (115) and 92% (241). Similar success rates were achieved with simple partial status epilepticus (163,241). As noted above, the VA Cooperative Status Epilepticus Trial demonstrated superiority of lorazepam (0.1 mg/kg) over phenytoin (18 mg/kg) in response rate to initial therapy (64.9% vs. 43.6%), with slightly better results for lorazepam than diazepam (0.15 mg/kg) (137). Intravenous lorazepam (4 mg) was effective against post–anoxic myoclonic status epilepticus after cardiac arrest in six patients (247). However, continuous EEG monitoring during lorazepam treatment is advisable, as electroclinical dissociation (electrographic seizures despite cessation of convulsive activity) has been observed.
Pediatric Status Epilepticus.
The usual IV lorazepam dose in pediatric status epilepticus is 0.05 mg/kg, repeated twice at intervals of 15 to 20 minutes (138,163). This regimen terminated seizure activity in 81% of 31 children aged 2 to 18 (241). In a prospective randomized trial of 178 children, lorazepam (0.1 mg/kg) had the same efficacy (100%) as diazepam (0.2 mg/kg) plus phenytoin (18 mg/kg), with similar rates of respiratory depression (about 5%) (248). A retrospective study found that lorazepam (0.1 mg/kg in children and 0.07 mg/kg in adolescents) was most effective in partial status epilepticus, terminating seizures in 90% of cases (163). Prior treatment of status epilepticus with phenytoin, phenobarbital, or diazepam did not alter the effectiveness of lorazepam, though chronic BZ treatment with clonazepam or clorazepate significantly reduced the effectiveness of lorazepam in status epilepticus (163), indicating tolerance. Respiratory depression, when observed, occurred after the first injection.
Lorazepam was effective in neonatal seizures refractory to phenobarbital and/or phenytoin in several small studies. In seven neonates (gestational ages 30 to 43 weeks) treated with IV lorazepam (0.05 mg/kg), seizures were controlled within 5 minutes in all seven patients, with no recurrence in five, and at least 8 hours of control in the remaining two patients (249). No respiratory depression or other adverse effects were reported. In another small series, status epilepticus in six of seven neonates was terminated with lorazepam (0.05 to 0.14 mg/kg) (250).
Pediatric Serial Seizures.
Sublingual lorazepam (1 to 4 mg) was effective against serial seizures in 80% (8/10) and partially effective in 20% (2/10) of children, with onset of clinical effect within 15 minutes in most cases (251).
Alcohol Withdrawal Seizures.
Lorazepam (2 mg) administered after a witnessed ethanol withdrawal seizure prevented a second seizure better than placebo (3% vs. 24%) and may be the agent of choice in this setting (252). Duration of withdrawal symptoms was shorter with lorazepam than with chlordiazepoxide (253).
Chronic Epilepsy.
Lorazepam was effective as adjunctive treatment of complex partial seizures, with an optimal dose of 5 mg/day after slow upward titration from 1 mg twice daily (189). Therapeutic levels were in the range of 20 to 30 ng/mL. However, long-term treatment with lorazepam is likely to result in tolerance and is not generally recommended (254).
Midazolam
Midazolam (see Fig. 50.1) is widely used for induction of anesthesia and as a preanesthetic agent. It is three to four times as potent as diazepam. Midazolam has gained popularity in acute treatment of status epilepticus by either IV or IM use, though its short duration of action necessitates continuous IV maintenance or subsequent therapy with an additional anticonvulsant. Midazolam (10 mg) IM injection reduced interictal spike frequency in EEG recordings as well as IV diazepam (20 mg) (255), and this route provides a valuable alternative when IV access is unavailable.
Pharmacokinetics
Midazolam is water soluble, but at physiologic pH, a conformational change in the BZ ring makes it lipid soluble (256). Serum midazolam levels after IV administration were best fit by a two-compartment model, with an initial tissue distribution phase (t1/2α of 5.7 minutes) and an elimination phase (t1/2β of 1.9 hour) (200). After IV administration in eight healthy adult volunteers, mean plasma concentration for a half-maximal increase in β-frequency activity on EEG recording was 276 μmol/L (257). With an IM injection, peak serum concentration occurred after 25 minutes (258). After oral administration, 44% of the dose was bioavailable (200), while intranasal midazolam bioavailability ranged from 50% (259) to 83% (260). Bioavailability after rectal administration was 52% (261) and 74.5% after buccal administration (102). Midazolam is 95% protein bound, with a volume of distribution of 1.1 L/kg and a half-life in the range of 1.9 (200,262) to 2.8 hours (258). The clearance rate was 6.6 mL/min/kg, with 56% urinary excretion. The pharmacokinetics of midazolam is altered in children and critically ill patients. In children aged 1 to 5 years, administration of midazolam (0.2 mg/kg) by intranasal or IV route resulted in a similar elimination half-life, 2.2 hours for intranasal and 2.4 hours for IV administration (263). In critically ill neonates, the elimination half-life after IV administration was 12.0 hours (264). In adult ICU patients, the volume of distribution (3.1 L/kg) and elimination half-life (5.4 hours) were significantly greater than in healthy volunteers (0.9 L/kg and 2.3 hours, respectively) (265) though clearance was not significantly different (6.3 vs. 4.9 mL/min/kg for patients and volunteers, respectively).
Midazolam is metabolized rapidly by α-hydroxylation of the methyl group on the fused imidazo ring (Figs. 50.1 and 50.5) (256). This metabolite is biologically active but is eliminated by hepatic glucuronidation, with an elimination half-life of about 1 hour (266).
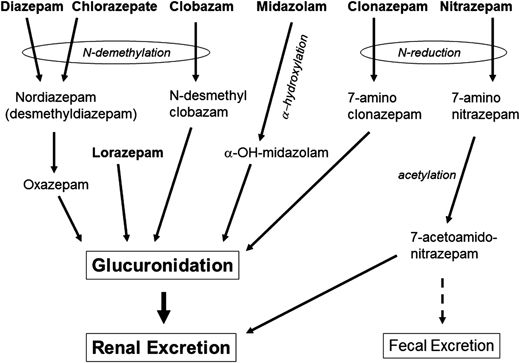
Figure 50.5. Hepatic metabolism of the antiseizure BZs.
Related posts:
 The Natural History of Seizures
The Natural History of Seizures
 Hormones, Catamenial Epilepsy, Sexual Function, and Reproductive and Bone Health in Epilepsy
Hormones, Catamenial Epilepsy, Sexual Function, and Reproductive and Bone Health in Epilepsy
 Epilepsy in the Setting of Inherited Metabolic and Mitochondrial Disorders
Epilepsy in the Setting of Inherited Metabolic and Mitochondrial Disorders
 Phenobarbital and Primidone
Phenobarbital and Primidone
 Outcome and Complications of Epilepsy Surgery
Outcome and Complications of Epilepsy Surgery
 Generalized Motor Seizures
Generalized Motor Seizures
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


