Chapter 13 Brain Plasticity and its Disorders
Introduction
Brain plasticity is an important concept that plays a major role in the expression of many pediatric neurological disorders and strongly influences recovery from brain injuries in neonates, infants, and children. Plasticity refers to the brain’s ability to change in response to experience, whether it is a positive experience such as education or practicing a skill, or an adverse event such as a stroke or other type of brain injury [Johnston, 2004]. The child’s brain exhibits greater plasticity than the adult brain, and common examples of enhanced brain plasticity in children include their ability to learn new motor tasks quickly, such as playing a musical instrument, participating in a sport, or their ability to become fluent in a new language [Meltzoff et al., 2009]. Children and adolescents are also able to form memories more easily than adults and they recover more quickly from brain injuries. The mechanisms responsible for various kinds of brain plasticity are being uncovered at a rapid pace as are the defects in these steps that cause intellectual disability and other pediatric disorders [Johnston, 2009]. This chapter gives a brief overview of normal brain plasticity and its disorders that are relevant to pediatric neurology, and provides insight into the pathogenesis of a variety of disorders described in other sections.
It is useful to consider four aspects or types of plasticity in the developing brain in order to understand how this concept is integrated into many disorders of the child’s nervous system (Box 13-1). The first type is generally referred to as adaptive plasticity, in which the nervous system changes in the process of learning new skills that have an adaptive advantage, such as learning to read or pass a test, learning to play a musical instrument, or learning to play a sport. Adaptive plasticity is also engaged when children recover from a brain injury, such as a stroke or extensive removal of brain tissue to cure epilepsy, and therapists try to harness this form of plasticity when providing speech, occupational, or physical therapy for a variety of disorders. In these cases, it is expected that normal childhood activities, such as attending school, music lessons, athletic practice, or therapies for cerebral palsy or other brain injuries, will activate normal plasticity programs to provide a good outcome that is beneficial for the child [Meltzoff et al., 2009]. Adaptive plasticity is generally enhanced in younger children compared with adults, and the genetically determined programs responsible for this kind of plasticity are heavily influenced by sensory input. Since learning and memory, as well as acquisition of physical skills, are very important for normal childhood development, genetic or acquired defects in the signaling pathways responsible for plasticity are reflected in a variety of developmental disability phenotypes [Johnston, 2004]. These disorders are best understood as examples of impaired plasticity. Many types of intellectual disability are caused by genetic lesions in the signaling cascades that are normally responsible for activity-dependent synaptic plasticity (Table 13-1). For example, in fragile X syndrome (FraX), the most common form of inherited intellectual disability, a trinucleotide repeat in the gene for the fragile X mental retardation protein (FMRP) leads to its absence or reduction in the brain [Penagarikano et al., 2007]. FMRP is responsible for transport of certain messenger RNAs and translation of proteins within dendrites in response to synaptic activity, and its absence causes a reduction in long-term potentiation (LTP), a physiological form of synaptic plasticity involved in learning and memory [Huber et al., 2002]. Accordingly, the intellectual disability and other behavioral features of FraX can be considered to reflect a genetically determined form of impaired plasticity in synapses. Therapies aimed at restoring normal plasticity mechanisms to synapses in FraX show some promise in preclinical and small clinical trials [Wang et al., 2010].
Box 13-1 Major Types of Plasticity in the Developing Brain




Table 13-1 Pediatric Disorders Caused by Genetic Mutations of Signaling Pathways Involved in Neuronal Plasticity
| Disorder | Mechanism of Disease |
|---|---|
| Neurofibromatosis 1 | Ras too active, enhanced GABA activity |
| Tuberous sclerosis | Upregulated mTOR signaling pathway |
| Rett’s syndrome | Mutations in MeCP2 transcription factor |
| Fragile X syndrome | Upregulated mGluR5 causes LTD |
| Coffin–Lowry syndrome | Mutations in RSK2 in Ras-MAPK pathway |
| Rubinstein–Taybi syndrome | Mutation in CREB binding protein (CBP) |
| X-linked intellectual disabilities | Mutation in PAK3 kinase: links RhoGTPases to dendritic cytoskeleton Mutation in oligophrenin, RhoGTPase Mutation in GluR3 AMPA receptor subunit |
| Costello’s syndrome | Upregulated H-Ras signaling to MAPK (intellectual disability, heart, skeletal disorder) |
| Lead poisoning | Enhanced PKC activity, inhibited NMDA receptors; impairs maturation of dendritic spines |
AMPA, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid; CREB, cyclic adenosine monophosphate response element binding protein; GABA, gamma-aminobutyric acid; MAPK, Ras-mitogen activated protein kinase; mTOR, mammalian target of rapamycin; NMDA, N-methyl-d-aspartate; PKC, protein kinase C.
Just as impaired plasticity is responsible for numerous brain disorders in children, maladaptive or excessive plasticity also can cause problems. When this phenomenon occurs, plasticity is misdirected or enhanced in a way that is harmful, or at least not beneficial. For example, children who have had strokes, traumatic brain injuries, or hypoxic-ischemic brain injuries sometimes develop delayed seizures that may result from formation of abnormal neuronal networks in damaged brain tissue [Kadam and Dudek, 2007]. In some cases, neurons in these networks have been shown to have functional changes in voltage-sensitive sodium channels that are similar to those found in certain forms of genetic epilepsy [Graef and Godwin, 2010]. Excessive plasticity may also be responsible for seizures and cognitive impairment in genetic disorders in which intracellular signaling pathways are enhanced, such as tuberous sclerosis and Costello’s syndrome [Crino, 2010; Dileone et al., 2010]. Another example of enhanced, but maladaptive, plasticity that causes neurologic impairment is dystonia in the hand and fingers resulting from over-practice of the piano or another musical instrument requiring intense finger movement [Quartarone et al., 2006]. In patients with this disorder, functional imaging of the contralateral cortex has shown that the somatosensory map of the fingers is blurred in the musicians with dystonia compared to controls [Elbert et al., 1998]. This suggests that the somatosensory cortex is less able to distinguish precisely between individual finger movements, and an attempt to use the fingers leads to dystonic movements instead. Interestingly, selective injection of botulinum toxin into some of the fingers can compensate for the disrupted somatotopic map of the fingers and restore normal movement [Cole et al., 1995]. The immature brain may be especially susceptible to organizational changes in neuronal circuits that lead to acquired disorders associated with maladaptive or excessive plasticity.
A fourth aspect of plasticity that it is important to consider is its potential to create cell-specific selective vulnerabilities, leading to the understanding of plasticity as the brain’s Achilles’ heel. This concept is especially important in the developing brain, where different cells are undergoing dramatic shifts in their composition during growth and development. One example is the subplate neurons that are among the first to reach the cerebral cortex in the second trimester of pregnancy, providing targets for axons projected from neurons in the thalamus [McQuillen et al., 2003]. These neurons are selectively vulnerable to hypoxia-ischemia due to their enhanced expression of excitatory α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA)-type glutamate receptors during that time period [Nguyen and McQuillen, 2010]. Immature oligodendroglia are similarly more vulnerable to injury during the second trimester due to the subunit composition of their AMPA receptors that favors permeability to calcium [Volpe, 2009]. N-methyl-d-aspartate (NMDA) receptors also contribute to developmental vulnerability of immature oligodendroglia [Salter and Fern, 2005]. These age-dependent changes in excitatory amino acid receptors on neurons and oligodendroglia that favor excitation have a beneficial role in normal development, but they can also make cells vulnerable to death if they are accidentally exposed to hypoxia or ischemia [McDonald and Johnston, 1990]. In a similar way, developing neurons in the brain closer to term become more vulnerable to excitotoxicity mediated by NMDA receptors due to the fact that the receptors are genetically programmed at that time to be more excitable [Monyer et al., 1993]. NMDA receptors are easier to activate in the developing brain due to their subunit composition, which makes it easier to open their channels. However, this characteristic also creates a vulnerability to injury if the brain is exposed to hypoxia-ischemia, which can result in neuronal depolarization and calcium flooding through opening of the NMDA channels.
In addition, gamma-aminobutyric acid (GABA), which normally has an inhibitory effect on neuronal circuits in older children and adults, mediates excitation in the newborn period because of developmental changes in chloride pumps in GABAergic synapses [Ben-Ari, 2006]. Enhanced activity of the NKCC1 chloride transporter in the neonatal period leads to higher intraneuronal concentrations of chloride than are present later on, so that, when GABA opens chloride channels, the cation leaves the neuron rather than entering [Dzhala et al., 2005]. Therefore, GABA leads to depolarization rather than hyperpolarization, as it does at older ages. Later in development, activity of the NKCC1 pump declines, and expression of the NKCC2 pump, which pushes chloride out of neurons, increases, leading to lower baseline concentrations of chloride inside the neuron compared to outside, and to inhibition. Combined with the expression of NMDA receptors, which are more active and flux more calcium and sodium in the fetal and neonatal brain, developmental changes in the actions of GABA lead to the brain being more excitable. This enhanced excitability during development appears to play an important role in the establishment of normal neuronal circuitry, which is dependent on electrical activity [Penn and Shatz, 1999]. For example, production of growth factors, such as brain-derived neurotropic factor (BDNF), occurs in neurons and is dependent on neuronal activity [Lessmann and Brigadski, 2009]. Prolonged blockade of NMDA receptors, just like too much activity, can cause neuronal death because a minimum baseline amount of channel opening, with entry of calcium and sodium into neurons, is essential for neuronal survival [Hansen et al., 2004]. On the other hand, this bias towards excitement in the developing brain is probably responsible for the higher propensity for seizures in infants and children compared to adults, and the expression of some seizure types such as infantile spasms [Hablitz and Lee, 1992]. Therefore it is quite important for the molecular machinery responsible for balancing excitement and inhibition in the developing brain to operate properly in order to prevent it from becoming an Achilles’ heel during periods of stress, such as hypoxia-ischemia or status epilepticus [McDonald and Johnston, 1990].
Basic Mechanisms for Plasticity in the Developing Brain
In addition to these four broad types of plasticity in the developing brain, there are at least six basic cellular mechanisms for plasticity (Box 13-2). The earliest mechanism is the overproduction of neurons and glia from stem cells, and then reduction of this population by apoptosis in the fetus [Haydar et al., 1999]. Most neurogenesis ceases after birth, but it continues in selected niches of stem-cell production in the subventricular zone of the lateral ventricles and the subgranular zone of the dentate gyrus of the hippocampus [Kernie and Parent, 2010]. These restricted zones of neurogenesis may contribute to recovery from brain injuries throughout life. Additional mechanisms for plasticity are:







Mechanisms of Synaptic Plasticity
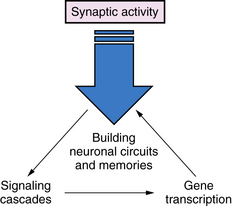
Synaptic plasticity is the most important mechanism for everyday activities such as learning and memory, and acquiring new skills (Figure 13-1). The strength of the signals between axons and dendrites across synapses can be increased or decreased through processes called long-term potentiation (LTP) or long-term depression (LTD), and these processes are strongly influenced by previous synaptic activity [Malenka and Nicoll, 1993]. The most prominent examples of LTP and LTD occur in excitatory glutamate synapses, which account for about 75 percent of all synapses in the brain. However, LTP and LTD are also present in many different types of neurons, including inhibitory GABAergic neurons and dopaminergic neurons [Nugent and Kauer, 2008]. One of the best examples of LTP is mediated by the NMDA-type glutamate receptors, which are voltage-dependent, meaning that NMDA channel opening is dependent in part on membrane depolarization [Gilland et al., 1998]. Channel opening is also dependent on occupancy of receptors for glutamate and glycine by these amino acids. When multiple excitatory axons depolarize the neuronal membrane, and glycine and glutamate occupy their receptors, the NMDA-activated calcium channel opens, allowing a pulse of calcium to enter the neurons and, in turn, activating a cascade of biochemical events that encode a memory of the event [Johnston, 2004]. The NMDA receptor has been referred to as a coincidence detector because it requires both membrane depolarization, stimulated by multiple presynaptic inputs, and activation of glutamate and glycine receptors to open its channel [Brown and Milner, 2003]. NMDA receptor-mediated LTP results in a “step up” in the signal mediated by the synapse during subsequent synapse activations, and it becomes easier to activate. Neuronal stimulation can also activate another type of a glutamate receptor, called a metabotropic gluR5 (mGluR5) receptor because it activates the second messenger phosphoinositol turnover, rather than opening an ion channel, as NMDA receptors do [Antion et al., 2008].
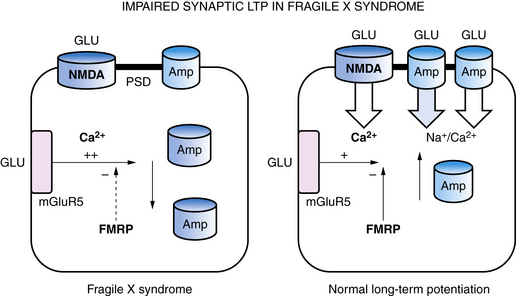
Within synapses, a variety of mechanisms contribute to LTP and LTD, including an increase or decrease in neurotransmitter release from presynaptic nerve terminals, a change in reuptake of neurotransmitter by astroglia that surround each synapse, a change in the number of neurotransmitter receptors in the postsynaptic membrane, changes in the flux of calcium and other ions through membrane channels, and an activation of intracellular signaling cascades [Citri and Malenka, 2008]. One of the common mechanisms that synapses use to increase or decrease synaptic strength is to alter the number of receptors in the postsynaptic membrane through a process called “receptor trafficking” (Figure 13-2) [Keifer and Zheng, 2010; Conboy and Sandi, 2010]. In this process, receptors shuttle back and forth between the cytoplasm, where they do not have access to neurotransmitter in the synaptic cleft, postsynaptic density, and postsynaptic membrane, where they are positioned to interact with neurotransmitter. One excitatory receptor that is heavily regulated by receptor trafficking in synapses is the AMPA receptor, which is the ionotropic glutamate receptor that carries most of the fast excitatory current in the brain. AMPA receptors shuttle between the dendritic cytoplasm, where they are inaccessible to glutamate, to the postsynaptic membrane, where they can be stimulated by glutamate [Keifer and Zheng, 2010]. The number of receptors within the synaptic membrane determines the strength of the synapse when it is activated. Accordingly, LTP is associated with an increase in AMPA receptors inserted into the postsynaptic membrane, while a decrease in AMPA receptors in the postsynaptic membrane is associated with LTD. LTP can be induced by rapid stimulation of the postsynaptic membrane, leading to voltage-dependent opening of NMDA receptors and trafficking of AMPA receptors from the cytoplasm into the postsynaptic membrane. This makes the synapse better able to conduct a signal, in effect creating a fragment of a memory about that synapse’s past experience. The mGluR5 metabotropic glutamate receptor also controls AMPA receptor trafficking, and an increase in mGluR5 activity leads to internalization of AMPA receptors, LTD, and weaker synaptic strength. In experimental models of fragile X syndrome, the activity of mGluR5 receptors has been reported to be increased, along with enhanced synaptic LTD, and antagonists of the mGluR5 receptor are able to restore LTP and improve abnormal behaviors and cognition (see Figure 13-2) [Muddashetty et al., 2007]. Fragile X syndrome is one example of a developmental brain disorder that can be understood at the level of the synapse. Synapses are thought to be the primary site of memory storage across large networks in which the strength of individual synapses is increased or decreased by the processes of LTP or LDP.
Production of nerve growth factors like BDNF is another mechanism by which previous synaptic activity can lead to plasticity in neuronal connections. BDNF is produced in neurons in response to neuronal activity, and it has been shown to increase and stabilize synaptic connections, as well as contribute to their maturation and integration into complex neuronal circuits [Yoshii and Constantine-Paton, 2010]. Learning has also been shown to increase neurotrophin signaling through the TrκB receptor, which mediates the actions of BDNF in the hippocampus in mice. BDNF has also been associated with cortical plasticity in humans. Kleim et al. reported that individuals with a val66met polymorphism (i.e., methionine codon substitution for valine codon at position 66 in the BDNF gene) showed reduced ability to reorganize their cortical motor map in response to a training exercise involving the fingers of one hand [Kleim et al., 2006]. In vitro studies showed that this mutation impairs the secretion of BDNF from neurons, but its expression is normal. McHughen et al. used functional magnetic resonance imaging (fMRI) to show that subjects with this BDNF mutation made more errors and had poorer retention on a driver-based learning task, and had altered short-term plasticity [McHughen et al., 2010]. BDNF genotype has also been shown to be associated with anxiety and memory problems, as well as with impaired functional connectivity from the hippocampus to the amygdala, insula, and striatal regions in children studied with functional connectivity (FC) analysis on data obtained through whole-brain fMRI [Thomason et al., 2009].
Plasticity of Dendrites and Dendritic Spines
Activity-dependent plasticity in excitatory synapses is associated with physical changes in dendritic spines that are thought to reflect information storage (Figure 13-3) [Hotulainen and Hoogenraad, 2010]. Immature dendritic spines are long with a small head, but repeated activity during development, including LTP, leads to shortening of spine shafts and enlargement of spine heads. New learning in animals also has been shown to be associated with localized enlargement of some dendritic spines and loss of others. Remodeling of dendrite morphology occurs continuously in association with synaptic plasticity, and is mediated by the actin-rich cytoskeleton that fills dendrites [Hotulainen and Hoogenraad, 2010]. The actin cytoskeleton within spines containing excitatory synapses provides support for the postsynaptic density (PSD), which anchors glutamate receptors and cell adhesion molecules that span the synaptic cleft (see Figure 13-3). NMDA receptors within the PSD regulate actin signaling pathways within an endocytic zone just below the PSD. The purpose of this zone is to recycle the synaptic pool of AMPA-type glutamate receptors involved in trafficking between the cytoplasm and the PSD. Therefore, the actin cytoskeleton plays an important role in LTP, and it is not surprising that mutations in proteins involved in regulation of this cytoskeleton, such as small Rho and Ras GTPases, alter spine morphology and cause cognitive disorders and autistic spectrum disorders [Pinto et al., 2010]. Similarly, key proteins that make up the PSD and the scaffolding proteins beneath the PSD, such as SHANK and the cell adhesion proteins – neurexins, neuroligins, and cadherins – are associated with clinical disorders that impair learning (Table 13-2) [Durand et al., 2007; Johnston et al., 2001].
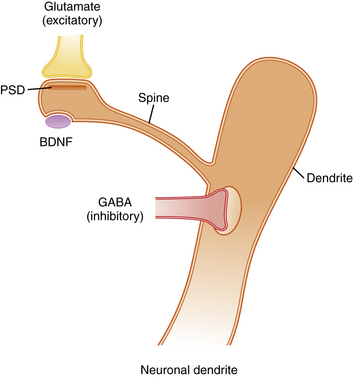
Excitatory axons that contain glutamate synapse on the tips of dendritic spines, while inhibitory axons that contain gamma-aminobutyric acid (GABA) synapse on the shafts of dendrites. Excitatory synapses are asymmetric and contain a postsynaptic density (PSD), which anchors neurotransmitter receptors and a variety of other proteins that provide scaffolding for receptors (see Table 13-2). Typically, each dendritic spine contains only one excitatory synapse, while the shaft of the dendrite contains multiple inhibitory GABAergic synapses. This arrangement allows a single dendrite to receive and process information from many excitatory synapses on its dendritic spines. However, the summed output for each dendrite is controlled by inhibitory receptors on the shaft. The morphology of dendritic spines is controlled in part by age and activity, so that young spines are long and thin with narrow spine heads, while older spines that have experienced considerable activity become shorter with a broader spine head. These shape changes are mediated by an actin cytoskeleton that fills the spines, allowing them to change shape in response to synaptic activity. Abnormal morphology of dendrites, spines, and PSD proteins is commonly associated with syndromes that include intellectual disability, autism, and behavior disorders. BDNF, brain-derived neurotropic factor.

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


