CHAPTER 97 Brain Tumor Immunology and Immunotherapy
An Immune Primer
The function of the immune system is to protect the body.1 Immunotherapy involves the administration of an agent to an individual that stimulates the immune system to react against something foreign or harmful, such as a tumor or an infection. The end result of a vaccine is to develop protective immunity against the foreign agent or tumor. This defensive function is performed by leukocytes (white blood cells) and a number of accessory cells distributed throughout the body. Lymphocytes are the key cells controlling the immune response. They specifically recognize foreign material and distinguish it from the body’s “self” components. Immunity may be cell mediated (T cells, natural killer [NK] cells, and phagocytes) or humoral (B cells, antibody, complement). When stimulated, cytokines are a group of molecules, other than antibodies, produced by lymphocytes that are involved in regulating the immune system. They include the interleukins, the interferons, tumor necrosis factor (TNF), and colony-stimulating factor (CSF). There are two main types of lymphocytes: B cells, which produce antibodies; and T cells, which have a number of functions including helping B cells to make antibodies (CD4 helper T cells, TH2) or helping cytotoxic T-cell responses (CD4 helper T cells, TH1); recognizing and destroying virus-infected cells (CD8 effector T cells); and controlling the level and quality of the immune response (regulatory T cells).
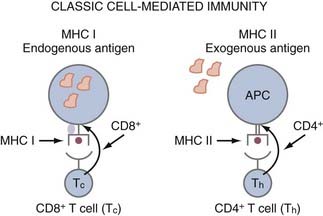
APCs are a group of cells that are capable of taking up antigens, partially degrading them, and presenting them to T cells in a form they can recognize. Although B cells recognize antigen in its native form, T cells only recognize antigenic peptide derivatives of complex antigens that have become associated with major histocompatibility complex (MHC) molecules. Thus, MHC molecules present antigen (i.e., peptides) to T cells. MHC class I molecules are found on all nucleated cells and platelets. MHC class II molecules (Ia antigens) required for helping B cells or making antibodies are expressed on B cells, macrophages, monocytes, APCs, and some T cells. CD8 cells (cytotoxic T lymphocytes [CTLs], killer) are class I restricted, meaning they only recognize antigen presented in the context of MHC class I molecules, whereas CD4 (helper T) cells are MHC class II restricted (Fig. 97-1). Antigens synthesized within a cell, such as viral polypeptides, associate preferentially with MHC class I molecules and present antigen directly to CD8 cells (“direct pathway”). In contrast, antigens that are taken up by an APC and partially degraded (processed) and returned to the cell surface associated with MHC class II molecules are recognized by CD4 cells (“indirect pathway”) (see Fig. 97-1). Syngeneic implies the same MHC as the organism or patient, and allogeneic implies a different MHC; hence the term allograft, which is rejection due to differences in MHC.
T cells in the CNS of healthy humans are a rare finding. However, during inflammatory responses, T cells are evident within the CNS. T cells require activation before entry into the CNS,2 but antigen specificity is not necessary for entry. T-cell infiltrates are commonly identified within human gliomas,3 and multiple studies have attempted to correlate the intensity of infiltration with survival,3–5 but this has not been consistently seen.6 One should bear in mind that these types of immunohistochemical assays do not take into account the functional activity of these cells or the contribution of inhibitor factors such as T regs. Thus, although these T cells have been activated in the systemic circulation, their functional activity has likely become impaired on entry into the tumor microenvironment,7 and it is not surprising that their presence in the tumor is not a definitive prognostic marker.
One concern about the use of immunotherapy in glioma patients is the induction of fatal autoimmunity. However, in previous immunotherapy trials of humans with brain tumors, there were only two possible cases of autoimmunity,8,9 so this remains a consideration but is likely related to the use of strong adjuvants.10 The rationale for selecting a tumor-specific antigen approach in immunotherapy is based on the facts that the targeting antigen preparation does not contain CNS antigens capable of inducing an autoimmune response and that the tumor-specific immune response can be monitored. However, the disadvantage of using a tumor-specific antigen is secondary to the heterogeneity of gliomas—one single antigen is unlikely to produce a durable response because a clonal negative population is likely to arise.11
It is widely recognized that tumors evade the host immune response through a number of mechanisms, including the elaboration of immunosuppressive cytokines, the production of immunosuppressive immune cells (T regs), and signal transduction disruption (Fig. 97-2).12
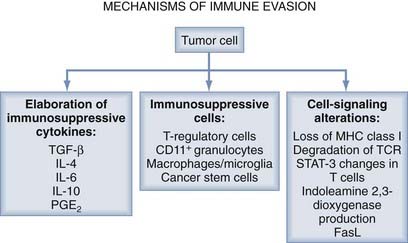
Mechanisms of Immune Suppression and Therapeutic Agents
Overview: Mechanisms of Immune Evasion
The secretion of immunosuppressive factors by the tumors themselves has recently been identified as a further target for immunotherapy. In particular, interleukin-10 (IL-10) and transforming growth factor-β (TGF-β) are two examples of such immunosuppressive cytokines secreted by tumor cells. Furthermore, there is increased expression of FasL and galectin by glioma that triggers T-cell depletion and T-cell apoptosis.13
Recently, a subset of regulatory T cells (T regs) have been identified in the local tumor environment and in the draining lymph nodes of systemic tumors, and they are elevated in the CD4 compartment of the systemic blood circulation. These cells coexpress CD4 or CD25, or both, and display potent immunosuppressive T-cell activity.14 In vaccines used for tumors outside the CNS, monoclonal antibody inhibition of these T regs has been associated with enhanced antitumor immunogenicity (details provided later). The inhibitory effects of these suppressor cells may also be mediated by cytokines. Interestingly, IL-2–mediated immune regulation involves not only the stimulation of effector cells but also the regulation of T regs.15
Regulatory T Cells
T regs inhibit T-cell activation and effector function by downregulating IL-2 production, inhibiting interferon-γ (IFN- γ) production, and increasing TH2 cytokine production,16 thereby inhibiting the expression of MHC class II molecules, CD40, CD80, and CD86 and suppressing antigen presentation by monocytes and macrophages.17,18 The presence of T reg–mediated suppression has been documented in a variety of human cancers.19 Several studies on immunogenic murine tumors have shown that adoptively transferred T regs are responsible for inhibition of tumor-reactive effector T cells and that the elimination of T regs enhances antitumor immunity.16 Within glioblastoma multiforme (GBM) patients, although reduced in absolute number, T regs comprise a disproportionately large fraction of the patient’s peripheral CD4 T-cell compartment. This increase corresponds with a decrease in T-cell effector functions. Furthermore, in vitro depletion of peripheral blood T reg cells results in successful reversal of effector T-cell function, including increased proliferation and a switch to a TH1 (IL-2–positive, tumor necrosis factor-α [TNF-α]–positive, IFN-γ–positive) cytokine profile. Within murine tumor challenge models of established gliomas, in vivo depletion of CD4+ and CD25+ cells resulted in significant long-term survival.20,21
Within the glioma microenvironment, the host-generated effector T cells can be critically suppressed or overwhelmed by the T reg cells. Tissues from glioma patients obtained after surgical resection have been dissociated and stained for the CD8+ and CD4+ subsets. The tumor-infiltrating CD8+ T cells were phenotypically CD8+ and CD25−, indicating that these cells were not activated or proliferating. CD4+ T cells were more numerous than CD8+ T cells within glioma tissue and the majority of CD4+ T cells were T regs, as evident by positive intracellular staining of FoxP3, a transcription factor that is elevated only in T regs and not in activated effector CD4+ T cells.22 These data indicate that T regs not only can inhibit initial systemic immune activation but also can prevent the effector responses in the tumor microenvironment. Of note, lower grade gliomas have only rare T regs, and oligodendrogliomas none at all. Even within the GBM patients’ specimens, the presence of T reg infiltration is highly variable and not an independent predictor of survival, emphasizing the redundancy of immunosuppressive pathways. Human T regs have been shown to accumulate in tumors and in ascites induced by the tumor and macrophages by the chemokine CCL22, which mediates trafficking of T regs to the tumor.19 Similarly, gliomas elaborate the chemokine CCL2, which induced the intratumoral migration of T regs.23
Regulatory T-Cell Inhibition by Chemotherapeutic Agents
Overcoming T reg immunosuppression is an ongoing immune therapeutic approach that can be achieved through a variety of approaches, including the use of denileukin diftitox (Ontak),24 cyclophosamide,25 or signal transducer and activator of transcription type 3 (STAT-3) blockade agents,26 or by inhibiting the T reg trafficking (i.e., inhibition of CCL2) with temozolomide.27 Inactivation of T regs with an anti-CD25 antibody in murine glioma models has been shown to enhance vaccination-induced antitumor immune responses and to result in the eradication of intracerebral astrocytomas without induction of autoimmunity.28 Similar results were also obtained in this murine model system with systemic cytotoxic T-lymphocyte–associated protein type 4 (CTLA-4) blockade. The CTLA-4 blockade reversed the CD4+ T-cell deficit, similarly seen in malignant glioma patients, and normalized the ratio of T regs in tumor-bearing mice.29 Although eliminating the suppression of endogenous antitumor immune responses through the elimination of regulatory T cells may enhance tumor immune clearance, there is a potential risk for inducing autoimmunity; however, that was not found in the murine model. It is likely that the induction of TH17 responses, and not necessarily the inhibition of T regs, is the primary mechanism of inducing CNS autoimmunity.30,31 In patients with metastatic renal carcinoma, Ontak specifically depleted the T regs without inducing toxicity and significantly improved the stimulation of tumor-specific T-cell responses when those patients were stimulated with RNA-transfected DCs.32 Without vaccination, Ontak has been shown to be efficacious as a single agent in relapsed and refractory B-cell non-Hodgkin’s lymphoma33 and in cutaneous T-cell lymphoma.34 However, Ontak was not particularly effective in clearing the T reg cells in melanoma patients.35 No published studies to date have evaluated this approach in malignant glioma patients. Another set of potential T reg modulation includes agents that block or disrupt Notch signaling. Notch signaling has also been identified as playing a role in regulating the responses of T cells and can affect the differentiation of CD4+ T cells into T regs.36 However, it is unclear which members of the Notch family need to be blocked, and inhibitors are currently under development.
Lymphodepletion to augment immunologic responses has been described in both murine model systems37,38 and human cancer patients.39 These enhanced antitumor responses after lymphodepletion may be secondary to the removal of competition at the surface of APCs,40 enhanced availability of cytokines that augment T-cell activity (e.g., IL-7 and IL-15),41 or depletion of immune inhibitory T regs.42 Cyclophosphamide (CTX), an alkylating agent with a therapeutic effect against tumors at high doses, has preferential effects on inhibiting T regs at lower doses. CTX can abolish the function of CD4+, CD25+, FoxP3+ T cells and enhance cytotoxic T-cell responses.43 Pretreatment with CTX before antitumor vaccination results in the activation of CD8 tumor-specific T cells.44 When CTX is administered at subtumoricidal doses, there is improved immune response and tumor eradication of large established murine tumors (sarcoma) when treated in combination with IL-12. This combination enhances CD4, CD8 T cells and macrophage infiltration within the tumor and skews the immune responses to the TH1 phenotype.45 Similar findings have been made in the murine system treating established melanoma with IL-2, IL-1, and CTX.46 In addition to its induction of immunity to new antigens, CTX can also overcome tolerance. For example, the administration of CTX to mice bearing an established plasmacytoma resulted in the cure of 92% of mice, and further studies demonstrated that the cured mice rejected a subsequent tumor challenge.47 The mechanism appeared to involve both the generation of CD8+ cytotoxic T cells and the upregulation of B7-1 (costimulatory molecule) on tumor cells.48
Multiple clinical trials have demonstrated enhanced immune responses and improved clinical efficacy when CTX was administered before immunotherapy.49,50 Administration of CTX to patients with advanced melanoma and colorectal cancer resulted in enhanced delayed-type hypersensitivity (DTH) reactions and the development of antibodies.51 When CTX was administered to augment autologous melanoma vaccine in patients with metastatic melanoma, the inclusion of CTX resulted in enhanced DTH responses.52 Similar potentiation of immune responses was seen in metastatic breast cancer and renal cell carcinoma. CTX has also been used in a phase I clinical trial of glioma patients and demonstrated some evidence of clinical efficacy. Plautz and colleagues53 treated newly diagnosed glioma patients with a single dose of CTX before the administration of adoptively transferred T cells harvested from the patients’ lymph nodes. This study was conducted before our current understanding of the influence of CTX on T regs, and thus the contribution of the CTX to the clinical efficacy is unknown.
Nevertheless, when combining the different forms of treatment, all aspects of treatment must be taken into account. Different dosing schedules and methods of vaccination can abrogate the desired therapeutic response. The use of low-dose doxorubicin after vaccination with a granulocyte-macrophage colony-stimulating factor (GM-CSF)–secreting cancer cell vaccine in a CT26 colon carcinoma murine model showed a significant increase in CD8+ T-cell response compared with the use of vaccine alone. Yet, when low-dose CTX was administered after or concurrently with a GM-CSF–secreting cancer cell vaccine, the desired immune response was inhibited, and the result was equivalent to that seen in mice treated only with chemotherapy.54 Thus, with CTX administered before vaccination, the reduction of suppressive immunity increased the immunotherapy-mediated tumor clearance. Similar results were also obtained with paclitaxel.55
Another agent that is capable of suppressing T regs is temozolomide (TMZ), the standard of care for GBM. TMZ is an alkylating agent that causes cell death by inducing G2/M-phase cell cycle arrest and likely autophagy without apoptosis.56 TMZ can inhibit the proliferation of lymphocytes, but it also depletes T regs27,57 and the trafficking of T regs into the glioma microenvironment.23 This mechanism may prove beneficial in combination with vaccine therapy and was recently investigated in a phase II clinical trial (ACT II) in combination with PEP-3-KLH vaccination.
In murine models of established intracerebral tumors, the combination of IL-2 immunotherapy and intratumoral administration of bischloroethylnitrosourea (BCNU) polymers enhanced survival.58 The investigators suggested that the cytotoxic agents might increase the number of tumor peptide antigens released or abrogate tumor-derived T-cell suppressor factors, or both. When mice with established tumors (lymphoma) were treated with BCNU, they rejected secondary tumor challenges, indicating the induction of immunologic memory. Furthermore, mice treated with BCNU demonstrated high cytotoxic activity against tumor cells and low T-cell suppressor activity. Thus, although BCNU was directly tumoricidal, it is also possible that the BCNU inhibited the T regs, resulting in antitumor immunity.59 The combination of BCNU with the newer efficacious immunotherapies has not been explored in animal models or clinical trials.
Immuno suppressive Cytokines
Human glioblastoma cells secrete a variety of factors, such as prostaglandin E (PGE), IL-10, and TGF-β2 that suppress multiple immune functions. For example, TGF-β2 inhibits cytotoxic responses of T cells against tumor targets, PGE2 regulates the generation of immune responses, IL-10 downregulates MHC class II expression on monocytes and suppresses T-cell proliferation,60–62 and vascular endothelial growth factor (VEGF) inhibits the maturation of DCs and subsequently downregulates MHC class II molecules. Suppression of TGF-β with an antisense TGF-β has been shown to be efficacious in 9L and C6 glioma intracerebral models.53,63 Other attempts at TGF-β inhibition in murine models have included SD-208, a TGF-βRI kinase inhibitor, which slightly increased median survival,64 and Tranilast, a TGF-β1 antagonist, which resulted in a 50% reduction in intracranial tumor volume.65 A phase I/II clinical trial using an antisense oligodeoxynucleotide (AP 12009) targeted to suppress TGF-β2 in high-grade glioma patients demonstrated an increase in median survival after recurrence that exceeded the current data for chemotherapy.66 The success of this type of approach is dependent on the dependency of the glioma on this particular mechanism of immune suppression and is compounded by the fact that gliomas usually express a variety of immunosuppressive cytokines; the blockade of any one cytokine may not be expected to significantly impact the overall immunosuppressive milieu.
The taxanes, paclitaxel and docetaxel, bind to ß-tubulin, stabilizing microtubules, and resulting in G2/M-phase mitotic arrest. Paclitaxel can diminish the cytotoxicity of NK and T cells by interfering with IL-2–mediated activation. However, the best-known immunomodulatory effect of paclitaxel is its ability to trigger macrophages to secrete a variety of proinflammatory factors, such as IL-1ß, GM-CSF, tumor necrosis factor-α (TNF-α), and IL-12. This lipopolysaccharide-like activity enables paclitaxel, in the presence of a priming signal, to induce the tumoricidal activity of macrophages.67 To date, there have been no reports of combining paclitaxel treatment with cancer immunotherapy.
Clearly, further studies investigating the cytokine profile before chemotherapy are warranted to enhance our understanding of the endogenous immune response. For example, in patients with breast cancer that were evaluated before treatment with chemotherapy, there was a significant decrease in immune cells such as NK and LAK cells and immunostimulatory cytokines such as IL-2, GM-CSF, and interferon compared with healthy individuals. After the administration of docetaxel and paclitaxel, there was a significant increase in the number of immune cells and a decrease in PGE2 and TNF.68 Doxorubicin has also been shown to have immunomodulating effects, including increasing the activity of monocytes and increasing the secretion of IL-1 and IL-2.69
Immune Inhibitory Molecules
To initiate an immune response, T cells must receive two signals: one through the T-cell receptor (TCR), which recognizes antigen presented within the context of MHC; and the second through the costimulatory receptor CD28, which recognizes the costimulatory molecules CD80 and CD86 expressed on the surface of the APC. In response to antigen-specific stimulation, activated T cells produce IL-2 and proliferate. B7-H1 is a negative costimulatory molecule that inhibits CD4+ and CD8+ T-cell activation, can induce T-cell apoptosis,70,71 and is upregulated within the tumor microenvironment72 and specifically in gliomas.70 Blockade of B7-H1 with a monoclonal antibody can activate T cells, which are then able to inhibit tumor.73 This represents a novel mechanism by which glioma cells evade immune recognition and destruction. Blockade of B7-H1 with antibodies in combination with adoptive T-cell transfer has been shown to enhance cancer clearance73 and enhance myeloid DC-mediated immunotherapy72; however, whether sufficient quantities of these types of antibodies can be achieved in the CNS remains to be seen. T-cell anergy can also be induced if the inhibitory molecule CTLA-4 is present on the gliomas.74 CTLA-4 blockade has had some success in promoting antitumor T-cell responses in both preclinical and clinical settings,29,75 but it is not clear whether this approach would be successful against primary brain tumors or CNS metastasis.
HLA-G is a nonclassic MHC molecule with limited tissue distribution that has an immune regulatory function. Soluble HLA-G (sHLA-G) has been identified in the plasma of patients with melanoma, glioma, breast cancer, or ovarian cancer. sHLA-G can inhibit the functions of T and NK cells at high concentration76; thus, at levels within the tumor microenvironment, it would be anticipated that there is reduced immunologic responsiveness. Gene transfer of HLA-G1 or HLA-G5 into HLA-G–negative glioma cells (U87-MG) rendered these cells highly resistant to direct cytolytic clearance, inhibited T-cell proliferation, and prevented efficient priming of cytotoxic T cells. The inhibitory effects of HLA-G were directed against CD8 and CD4 T cells, but appeared to be NK-cell independent.77 Only a few HLA-G–positive cells within the tumor microenvironment are necessary to exert significant immune inhibitory effects77; thus, the inhibition of HLA-G blockade would need to be comprehensive. However, the frequency of expression of HLA-G and B7-H1 has not been assessed across glioma grades or types on tissue arrays; thus, the universality of this type of immune evasion is not currently known.
Phenomenon of Antigen-Loss Variants in Gliomas
Although tumor specificity has the advantage of not inducing potentially fatal autoimmunity, the intrinsic antigenic heterogeneity will prevent clearance of all cancer cells and the development of antigen-loss variants, especially for those antigens whose expression is not required by tumor cells for the maintenance of a transformed phenotype. The propagation of such antigen-loss variants is secondary to epitope immunodominance, that is, the preferential immunodetection of one or a few epitopes among many expressed on a given target.78 The parental tumor cells carrying the immune dominant epitope can be eliminated, and the formerly immune recessive epitopes then become dominant, allowing unrestricted tumor growth. It is well documented that tumor antigen expression is heterogeneous, even within the same tumor,79 and antigen loss has been observed with a variety of immune therapeutic approaches in various cancers.80,81
The epidermal growth factor receptor (EGFR) is often amplified and structurally rearranged in malignant gliomas, with the most common mutation being EGFRvIII. Treatment in a murine model with a vaccine consisting of a peptide encompassing the tumor-specific mutated segment of EGFRvIII (PEP-3) conjugated to keyhole limpet hemocyanin (KLH [PEP-3-KLH]) resulted in the loss of antigen expression. Specifically, EGFRvIII expression in mice that failed to respond to the PEP-3-KLH vaccination was lost in 80% of relapsing tumors, indicating that EGFRvIII-negative escape variants were a potential mechanism of treatment failure in active immunotherapy.11 Although EGFRvIII appears to represent a nearly terminal branch of tumor progression for malignant brain tumors and is expressed clonally within this lineage, antigen-specific tumor targeting might confer a selective growth advantage on neoplastic cells not expressing this epitope. It has been shown previously that treatment failure with antigen-specific passive humoral immunotherapy is not a result of antigen-loss variants,82 suggesting that antigen-negative escape variants are an etiology of treatment failure in active immunotherapy. In two separate phase II clinical trials (ACTIVATE and ACT II) in newly diagnosed EGFRvIII-expressing GBM patients who received the PEP-3-KLH, on tumor progression, the EGFRvIII expression was lost.57,84 To overcome this obstacle, cocktails of different antigens or whole-tumor lysates can be used as immunogens85; however, with nonspecific antigen approaches, there is a risk for autoimmune responses.10 To circumvent this, future approaches may involve screening the patient’s tumor for a panel of antigens86 and targeted immunotherapy selected based on these parameters—similar to selecting antibiotics for a given bacterial infection. Alternatively, upregulating the expression of these antigens with some forms of chemotherapy may restore the ability of the immune system to clear these cancer cells.87
A variety of agents have been shown to be capable of inducing or upregulating antigen expression. For example, gemcitabine, a nucleoside analogue, can increase tumor antigen cross-presentation and T-cell infiltration of the tumor.88 The demethylating agent DAC is capable of inducing the tumor-associated antigen MAGE-1 on melanoma cells with subsequent cytolytic T-cell clearance.89 Irinotecan (CPT-11) has been shown to induce the LYGD/E48 antigen, which resulted in synergistic tumor regression with a cytotoxin-armed antibody in a colorectal xenograft murine model.90 The combination of 5-fluorouracil and cisplatin (CDDP) can upregulate the antigen CEA and MHC class I in human colorectal cancer cell lines91 and was functionally relevant as evidenced by increased CEA-specific, MHC-restricted cytotoxic T-cell killing.87 Thus, many different types of chemotherapy can enhance antigen expression; however, none of these agents has been tested to determine whether it can restore antigen expression after clonal elimination with immunotherapy.
An alternative therapeutic approach for the issue of antigen heterogeneity would be to selectively screen each patient’s tumor for a panel of potential antigens, but this would be somewhat labor intensive and costly at present. Another alternative approach would be to vaccinate cancer patients with a panel of tumor-associated or tumor-specific antigens. Tissue arrays of gliomas from a large population of patients could be used to determine the frequency of the most common tumor antigens. This strategy would be very similar to that used in manufacturing flu vaccines annually, in which the most common virulent strains are incorporated as immunologic targets. Finally, exploration has begun on immunologic approaches for inducing immune responses against immune escape variants based on defects in the transporter associated with antigen processing,92 but these are not yet sufficiently developed for clinical trial application.
STAT-3—A Key Switch Mediating Immuno suppression
The ability of tumor cells to proliferate uncontrollably, resist apoptosis, sustain angiogenesis, and evade immune surveillance is regulated, in part, by the signal transducer and activator of transcription type 3 (STAT-3), which acts as a cytoplasmic signaling protein and nuclear transcription factor. Under normal physiologic conditions, latent STAT-3 activation is dependant on ligand-receptor interaction, primarily under the control of growth factor receptor tyrosine kinases, cytokines, and G-protein receptors.93 For example, EGFR and IL-6 activate Jak2, which then activates STAT-3 by phosphorylation of the tyrosine residue in the transactivation domain of STAT-3.94 In cancer cells, these tyrosine kinases are among the most frequently activated oncogenic proteins. STAT-3 has been shown to be persistently activated in most human cancers, including gliomas.95,96 The phosphorylation of STAT-3 has been attributed to EGFRvIII.97 IL-6 is expressed in the CNS, especially by reactive astrocytes, under a wide variety of conditions, such as hypoxia,98 traumatic and metabolic injury,99 and inflammation,100 which in turn also induces expression of p-STAT-3. The use of decoy antisense STAT-3 oligonucleotides and dominant-negative vectors has provided convincing evidence that STAT-3 is highly relevant to the growth and survival of many tumor types, including gliomas.101–103
STAT-3 has also emerged as a key switch that drives the underlying immuno suppression104 identified in cancer patients by preventing maturation of DCs and inhibiting the proliferation and activation of immune effector populations. STAT-3 has been shown to be a potent regulator of these anti-inflammatory responses by suppressing macrophage activation and limiting inflammatory responses.105,106 It also has been shown to become constitutively active in diverse tumor-infiltrating immune cells.107 For example, STAT-3 activity within NK cells and neutrophils directly reduces their cytotoxicity, whereas STAT-3 activity in DCs reduces the expression of MHC class II, CD80, CD86, and IL-12 in these cells, rendering them unable to stimulate T cells and generate antitumor immunity. Investigators have recently shown that by ablating STAT-3 in only the hematopoietic cells in mice resulted in marked enhancement of activated and functional T cells, NK cells, and DCs in tumor-bearing mice. This ablation of STAT-3 in only the hematopoietic cells also resulted in marked antitumor effects in vivo, indicating that STAT-3 expression in immune cells restrains antitumor immune eradication.107 The tumor microenvironment induces STAT-3 activity in tumor-associated immune cells.104,107,108
The blockade of STAT-3 with small molecular inhibitors in microglia obtained from the CNS of patients with GBM resulted in the upregulation of costimulatory molecules (e.g., CD80, CD86), induced systemic APCs to elaborate proinflammatory cytokines essential for T-cell effector responses, and induced activation and proliferation of T cells, indicating that STAT-3 blockade is a potent approach to modulating both the systemic and local tumor immune microenvironments. Of note, this potent immune activation occurred in immune cells obtained from patients with disease refractory to other conventional immune activators, such as toll-like receptor agonists. When the immune cells from immunosuppressed GBM patients were treated with a STAT-3 inhibitor, Western blot analysis demonstrated phosphorylation of ZAP-70 in T cells, thus bypassing the impairment of signal transduction typically identified in the immune systems of patients with cancer.109 The mechanism of T-cell activation is likely secondary to the inhibition of T regs present in these immune preparations, thus allowing immune activation (i.e., releasing the “brake” on immune cell activation and proliferation). IL-2 has been shown to regulate FoxP3 expression in human CD4+, CD25+ T regs by inducing STAT-3 binding of the first intron of the FoxP3 gene.110 Suppressor of cytokine signaling type 3 has been shown to be an inhibitor of STAT-3 signaling111 and transcriptional activity112,113 but is deficient in T regs.114 STAT-3 inhibitors should have a multiplicity of immune modulatory activities, such as allowance of expression of costimulatory molecules in the tumor microenvironment, inhibition of immune suppressive cytokines, maturation of DCs, and inhibition of T regs, and represents a newly emerging class of drugs that will be used for immune therapeutic purposes.
Immune Modulation by Central Nervous System Antigen-Presenting Cells
T-cell activation requires signals through both the MHC and costimulatory molecules, and the expression of MHC alone results in T-cell anergy.115 Low levels of expression of CD80 provide an immune escape advantage to cancer cells, and CD28-mediated costimulatory signals are essential for differentiation of functional tumor-specific cytotoxic CD8+ effector T cells.116,117 In tumor-bearing animals, repeated stimulation of the T cells is necessary, especially at the effector site, to generate tumor responses,118 indicating that if the APC failed to provide appropriate stimulation, the APC may contribute to immune suppression. Although microglia may be able to act as APCs during autoimmunity (multiple sclerosis) and infection (HIV),119,120 glioma-associated microglia are functionally impaired, are not able to stimulate T-cell responses, and are refractory to activation stimulation (i.e., toll-like receptor agonists).7 Intratumoral DCs may be able to provide these immune-stimulating conditions, but these are rarely present in malignant gliomas.7 Additionally, CD11b+ inhibitory macrophages (iMACs) have been described, which induce apoptotic death in CD8+ T cells. This inhibitory phenotype could be changed into one reflective of highly activated DCs by culturing the iMACs with IL-4 and GM-CSF,121 suggesting that the tumor microenvironment can be manipulated for therapeutic purposes. However, the delivery of cytokines directly to the CNS is problematic. Costimulatory molecules on glioma-associated microglia and macrophages have been shown to be lost or significantly downregulated. In attempts to overcome the failure of APCs, and specifically the lack of sufficient costimulation, vaccines with enhanced costimulatory capacity have been tested and demonstrated antitumor effects that have maintained high avidity effector-memory CD8+ T cells. Other approaches for potentially upregulating the costimulator molecules on APCs include CpG DNA,122 chemotherapy with cytosine arabinoside,123 transfection with poxviruses expressing the costimulatory molecules,124 or transfection of the replication-defective fowlpox recombinant vector rF-TRICOM (TrIad of COstimulatory Molecules).125
An emerging strategy for upregulating costimulatory molecules in the glioma microenvironment is with STAT-3 blockade agents. A small molecular inhibitor (WP1066) of STAT-3 has been shown to upregulate costimulatory molecules and proinflammatory cytokines on microglia obtained from the CNS of patients with GBM that were refractory to modulation by other conventional immune activators, such as toll-like receptor agonists. Furthermore, WP1066 can modulate enhanced T-cell functional activities in physiologic ranges that can be obtained within the CNS.109 These types of agents are anticipated to be in clinical trials of human patients within the next several years.
Immune Modulation by Glioma-Infiltrating Lymphocytes and TH2 Lymphocytes
After activation, CD4+ helper T cells are classified, based on their elaborated cytokine responses, as TH1 (promoting cell-mediated immunity) or TH2 (promoting humoral immunity), which predominantly secrete either interferon-γ (IFN-γ) or IL-4, respectively. A predominance of TH2-type cytokine response has been correlated with enhanced tumor growth.126,127 This TH2 polarization has been demonstrated in gliomas128 and is likely mediated by microglia-secreted macrophage-derived chemokine (MDC)129 or neuropeptides, or both.130 One therapeutic approach altering the TH2 intratumoral skewing would be to limit the chemokines that direct migration of the immunosuppressive immune cells to the tumor microenvironment, such as with a STAT-3 inhibitor.26 Alternatively, preferential TH1 responses could be induced with cyclooxygenase-2 (COX-2) inhibition.131
Impaired Immune Responses
The proliferative response of stimulated T cells, along with the levels of the IL-2 receptor and tyrosine phosphorylation in response to IL-2, from malignant glioma patients are reduced compared with healthy donors.132 These results suggest a defect in the expression of functional IL-2 receptor and T cells isolated directly from human gliomas that are not activated as reflected by expression of the IL-2 receptor CD25.7 Soluble products from tumors may suppress T-cell proliferation through a mechanism that involves downregulation of Jak3 expression and inhibition of IL-2–dependent signaling pathways.133 A potential mechanism for overcoming this is the constitutive activation of the T cells by transfection with CD3 or CD28, or both, for adoptive transfer therapeutic purposes. An antibody, r28M, directed against a melanoma-associated proteoglycan expressed on glioblastoma cells activates T cells through the CD28 molecule without additional stimulus from the TCR-CD3 complex and induces T-cell–mediated killing of glioblastoma cells in vitro and in vivo.134
Doxorubicin, an anthracycline antibiotic, can enhance the cytotoxic T-cell compartment. Pretreatment of mice with doxorubicin produced enhanced cytotoxic T-cell responses and activation of macrophages, as reflected by the secretion of IL-1 and IL-2.51 There was a twofold increase in the number of macrophages that could support the generation of the cytotoxic T cells. Ultimately, the macrophages became tumoricidal, and the effect persisted for 14 to 18 days. Further studies demonstrated that the administration of doxorubicin 5 days after a GM-CSF–secreting vaccine injection potently induced cytotoxic T-cell activity.54 Cancer patients treated with doxorubicin at a dose of 25 mg/m2 developed an increase in the percentage of circulating CD8+ T cells having twofold higher cytotoxic activity.135 Ideally, the chemotherapy selected for use in combination with immunotherapy should have some intrinsic tumoricidal activity. Although doxorubicin has little clinical efficacy against gliomas, this approach could potentially be used to boost the T-cell responses in glioma patients with further clinical development.
T-Cell Apoptosis
T-cell apoptosis has been observed frequently within malignant gliomas.136 The induction of apoptosis involves the activation of caspases, leading to nuclear fragmentation and apoptosis, and is triggered by deprivation of survival stimuli such as costimulators7 or by binding of ligands to death-inducing membrane receptors.137 Both of these mechanisms are operational within gliomas. FasL is predominantly expressed on T cells after activation by antigen and IL-2. When mature T cells are repeatedly stimulated by antigens, they coexpress Fas and FasL.137 Gliomas can evade killing by tumor-infiltrating T cells by overexpressing FasL, rendering the tumor resistant to apoptosis138 while delivering a death signal to Fas-expressing cells, which include the activated cancer-infiltrating T cells. However, high-grade gliomas also express the apoptotic receptors Fas and CD95, rendering them susceptible to antibody-stimulated Fas- and CD95-mediated apoptosis and CD8+ cytotoxic killing.139 Alternatively, agents such as topotecan can be administered to patients to upregulate Fas on gliomas that renders the tumor more susceptible to cytotoxic immune cell clearance.139–141
Topotecan penetrates the blood-brain barrier but has demonstrated only modest clinical efficacy in glioma patients, probably owing to failure to achieve in vivo levels sufficient to induce apoptosis of the tumor. Nevertheless, topotecan has been shown to upregulate Fas and CD95 in high-grade gliomas, rendering them more susceptible to immune clearance.139,141 Thus, although levels achieved are insufficient for direct glioma clearance within the CNS, this agent potentially could be exploited as an immune modulator. Other chemotherapeutics, such as doxorubicin142 (Adriamycin) and its analogues (epirubicin and pirarubicin),143,144 VP-16 (etoposide), cisplatin,144 and the combination of cisplatin and 5-fluorouracil145 can also increase cytotoxicity mediated by T cells and NK cells by upregulating Fas.
A synergistic antitumor effect was observed with paclitaxel and FasL in two human malignant glioma cell lines, T98G and LN-229.146 The synergy of paclitaxel and FasL was achieved independently of either the G2/M-phase cell cycle arrest or p53 activation. At low concentrations of paclitaxel, Bcl-2 phosphorylation was induced in the glioma cells, which in turn interfered with the heterodimerization of Bcl-2 with Bax and the inhibition of Fas-induced apoptosis by Bcl-2. The investigators hypothesized that the synergistic antiproliferative effect of paclitaxel and FasL on malignant glioma cells stemmed from the upregulation of the Bcl-2/Bax rheostat in favor of Bax, thereby sensitizing the tumor cells to apoptosis. Interestingly, this mechanistic synergy was not observed with other agents such as teniposide.147
T-cell apoptosis is also induced by glioma-expressed CD70 and gangliosides. The glucosylceramide synthase inhibitor (PPPP) has been shown to reduce the ability of GBM cell lines to induce apoptosis in T cells.148 Alternative strategies for blocking the T-cell death pathway include proteosome inhibitors, antisense oligonucleotide inhibitors, and other small molecule inhibitors. Although preclinical trials using some of these compounds have been successful in regressing established tumors, their therapeutic efficacy in cancer patients has yet to be established. Furthermore, apoptotic or necrotic tumor death is a desirable effect, and specific agents that block only T-cell apoptosis have yet to be devised.






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


