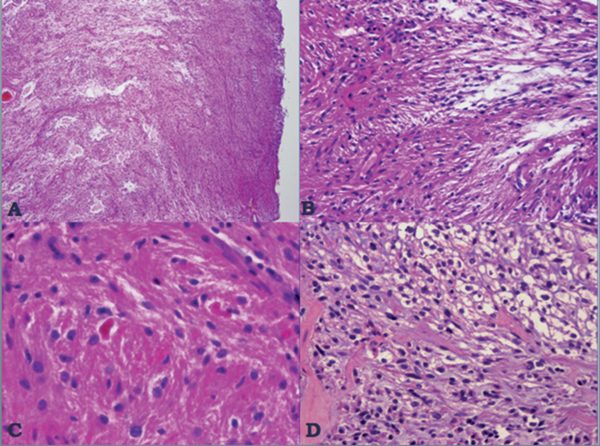
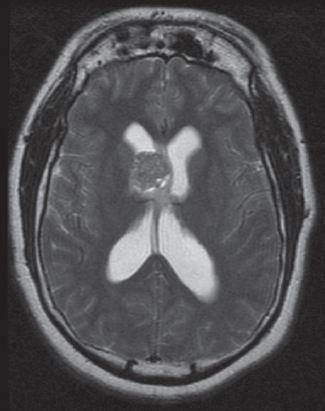
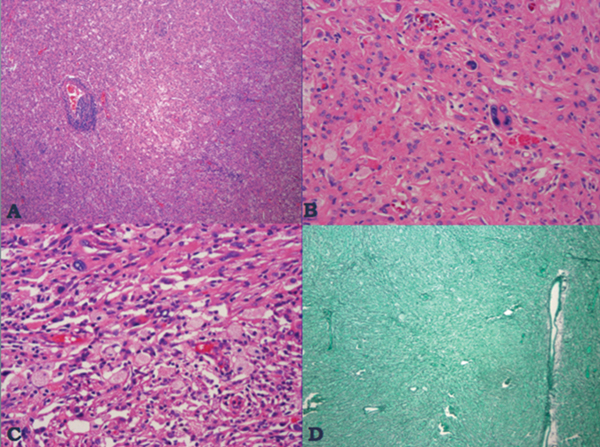
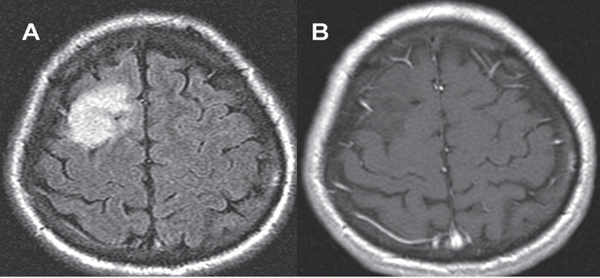
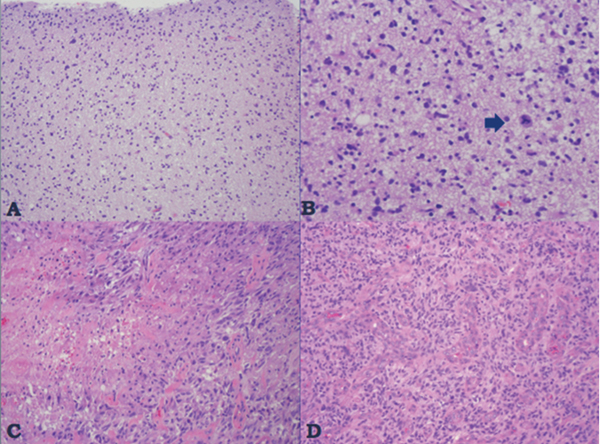
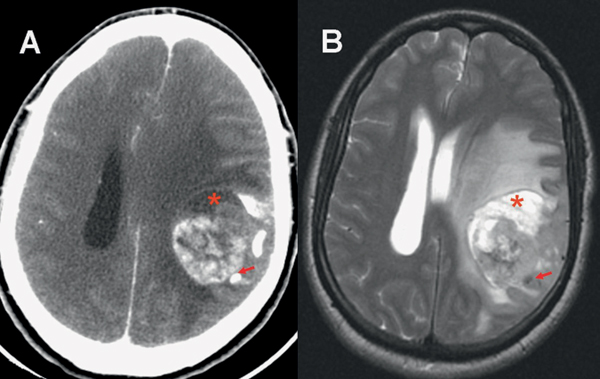
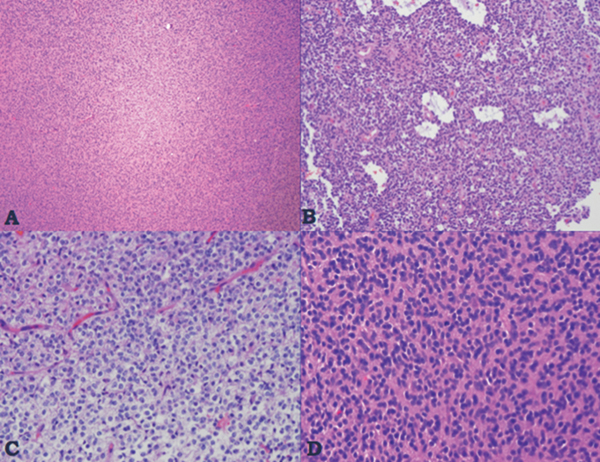
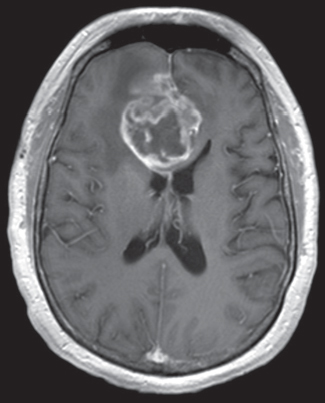
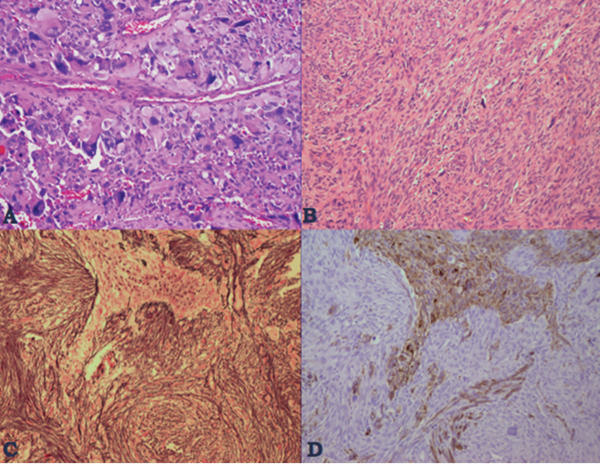
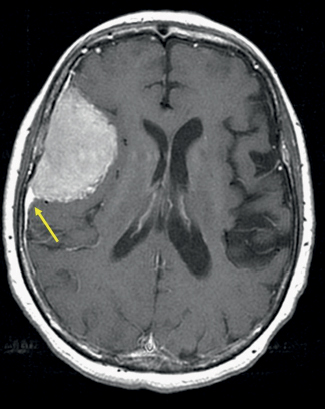
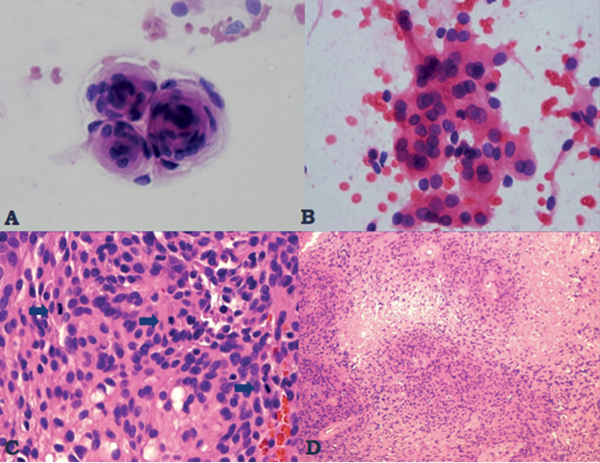
35 True or false: Most adults diagnosed with a low-grade glioma will not die of their brain tumor. False; low-grade gliomas should generally be considered an indolent but ultimately deadly disease. They are not benign. What is the typical presentation for an adult patient with a low-grade glioma? Normal adult with sudden onset of epilepsy in the absence of other focal neurological findings Are there any other possibilities? A small number (approximately 1%)1 present with signs of increased intracranial pressure (ICP) or a focal neurological deficit Approximately how long can an adult patient expect to live after having been diagnosed with a low-grade glioma? 10 years1 Roughly what percentage of low-grade gliomas ultimately progress to a higher grade lesion in adults? 50%1 What neuroimaging modality should be used to examine low-grade gliomas? MRI, because CT cannot delineate the extent of the tumor as well, and may even miss some small lesions altogether How do the lesions appear on MRI? Nonenhancing with a variable degree of infiltration What is the significance of enhancement? Microvascular proliferation is likely present, suspicious for high-grade glioma Are there any exceptions to this general rule? Yes; in pilocytic astrocytomas and pleomorphic xanthoastrocytomas, which present in young adults, enhancement is not necessarily an inauspicious sign. What kinds of glial cells may become gliomas? Astrocytes > oligodendrocytes > ependymal cells Name the four categories of low-grade gliomas. • Astrocytoma (grades I and II) • Oligodendroglioma • Mixed oligoastrocytoma • Miscellaneous (pleomorphic xanthoastrocytoma, ganglioglioma, and other glioneuronal tumors) How does the “miscellaneous” category differ from the other low-grade gliomas? These tumors all tend to have distinct radiological appearance and they may be completely resected (though serial follow-up is still indicated as they may recur). What chromosomal abnormalities are most strongly associated with low-grade astrocytomas? Loss of sex chromosome or p53 (17p) Which WHO grade I astrocytoma occurs most commonly in patients with neurofibromatosis type 1 (NF1)? Pilocytic astrocytoma How would you generally describe this tumor? Slow growing, well-circumscribed, variably enhancing grade I lesion True or false: Bilateral optic nerve involvement is a common presentation of this tumor. True. How do pilocytic astrocytomas appear radiologically? Well-circumscribed, with a variably enhancing solid component (94% enhance),1 either with microcysts or predominantly cystic with a mural nodule; the majority are periventricular2 Can you use CT to visualize this lesion? Yes; pilocytic astrocytoma will enhance on both CT and MRI. With what age group is this tumor associated? Children and young adults (first two decades of life)3 Where would this type of lesion most commonly appear in this age group? Cerebellum (67%)4 What are the common presenting signs and symptoms? Focal neurological deficits or nonlocalizing signs such as macrocephaly, headache, endocrinopathy, increased ICP Are seizures common? No, because the cerebral cortex is seldom involved What would you expect to find histologically? A biphasic (loose and compact) tissue pattern, with astrocytes that have elongated nuclei and thin bipolar processes. Rosenthal fibers, eosinophilic granular bodies, hyaline droplets, microcysts, and areas with regressive changes (degenerating nuclei, hyalinized blood vessels) may be seen. Mitotic figures are rare.5 Fig. 35.1 Pilocytic astrocytoma shows a biphasic pattern at low magnification (A), with areas showing cystic change. Piloid astrocytes show elongated nuclei and bipolar processes (B). Rosenthal fibers seen in compact areas (C). Areas may also show oligodendroglia-like morphology (D). Is the presence of Rosenthal fibers necessary and sufficient for diagnosis? No and no. Though they may be helpful in diagnosis, they are not required, and they may be found both in other neoplastic processes (ganglioglioma) and in chronic reactive gliosis. Are these lesions GFAP-positive? Yes What is meant by “pennies on a plate?” This phrase describes the circumferential location of nuclei in large/giant degenerating cells. True or false: p53 mutation and aberrant platelet-derived growth factor (PDGF) signaling play an important role in the development of pilocytic astrocytoma (PA). False; unlike diffuse astrocytomas, pilocytic lesions lack p53 involvement and instead exhibit increased immune response genes and aberrant neurogenesis. Recently, BRAF mutations have been strongly associated with PA. Are the genetics of NF1-associated and sporadic pilocytic astrocytomas the same? No. NF1-associated lesions show loss of normal NF1 expression and constitutive RAS (RAt Sarcoma protein) activation resulting in downstream mTOR (mammalian Target Of Rapamycin) hyperactivity. Sporadic lesions do not show loss of NF1 expression; they may even show NF1 hyperexpression. What is the prognosis with this lesion? More than 95% of patients have a 25-year survival rate if the enhancing portion of the tumor is totally resected. What type of change in the nature of these lesions is seen over time, if any? Regressive/degenerative rather than anaplastic change When, rarely, a pilocytic astrocytoma undergoes malignant transformation, is it considered a glioblastoma? No, even with malignant transformation, the prognosis is not necessarily poor, so the term anaplastic pilocytic astrocytoma is preferred. How does the variant pilomyxoid astrocytoma differ from pilocytic astrocytoma? • Grade II tumor • Histological differences • Age at presentation (younger in pilomyxoid) • No association with NF1 What are the specific histological differences? • Prominent myxoid/mucoid matrix • Monomorphous cells that are angiocentric (may resemble pseudorosettes) • Absence of Rosenthal fibers and eosinophilic granular bodies • Mitotic figures may be present At what age does this tumor typically present? Mean age 18 months, but it can occur in older children What is the prognostic difference? More likely than pilocytic astrocytoma to recur locally and/or have cerebrospinal spread6 Why is pilomyxoid astrocytoma (formerly known as “infantile pilocytic astrocytoma”) considered a variant of pilocytic astrocytoma? Because the tumor may occasionally phenotypically convert to a typical pilocytic astrocytoma What WHO grade I tumor is the most common neoplasm associated with tuberous sclerosis? Subependymal giant cell astrocytoma (SEGA); incidence of 6 to 14% in patients with tuberous sclerosis4 Fig. 35.2 MRI T2-weighted axial image demonstrating SEGA at the level of the foramen of Monro. Can SEGA occur without tuberous sclerosis? This is debatable. SEGA is one of the major diagnostic criteria for tuberous sclerosis.7 Can this tumor occur congenitally? Yes, though it most commonly occurs during the first two decades of life. How does it present clinically? Either with worsening epilepsy or signs of increased ICP (due to obstructive hydrocephalus) How does SEGA arise? From the subependymal hamartomas of tuberous sclerosis along the surface of the lateral ventricles Is SEGA GFAP-positive? Yes What “meningocerebral” grade II astrocytoma arises supratentorially in <90% of cases? Pleomorphic xanthoastrocytoma (PXA) (called “meningocerebral” due to its arising in the superficial cerebral cortex and often involving meninges)8 In which lobe is this neoplasm most commonly found? Temporal lobe In what age group are the majority of PXA cases found? <18 years of age; can also occur in adults between 62 and 82 years of age8 How do these patients tend to present? A long history of seizures To what does the term pleomorphic refer? The cells in this tumor have highly variable cytological features. What are some of the various cytological features that may be observed? Mono- or multinucleated giant astrocytes with abundant eosinophilic cytoplasm, admixed with spindle cells and xanthomatous cells. Eosinophilic granular bodies and perivascular lymphocyte aggregates are also frequent. What about “xanthoastrocytoma?” Large cells with abundant intracytoplasmic vacuoles due to lipid accumulation Fig. 35.3 Pleomorphic xanthoastrocytoma is a solid neoplasm, showing areas of xanthomatous cells as well as perivascular lymphoid aggregates (A). Large cells with multiple nuclei are seen (B), as are smaller spindle-shaped cells (C). Silver stain highlights the abundant pericellular deposition of reticulin fibers (D). What other finding is characteristic of PXA? • Pericellular deposition of reticulin fibers (visualized with silver stain) • Perivascular lymphocyte aggregates This feature is particularly helpful for differentiating it from what other neoplasm? Glioblastoma (GBM) PXA is immunoreactive for which three antibodies? S-100, GFAP, and synaptophysin Is there any association with hereditary tumor syndromes? PXA is weakly associated with NF1 Is p53 a major player in PXA? No. There has been little association with p53; the most common chromosomal alteration is −9 (50% of cases) What is the prognosis with this tumor? 80% survival at 5 years and 71% at 10 years8 What factors are most indicative of a favorable outcome? • Complete surgical resection • Low mitotic index What WHO grade II astrocytoma is characterized by a high degree of cellular differentiation and slow growth but commonly progresses to anaplastic astrocytoma and/or GBM? Diffuse astrocytoma What is another term commonly used for diffuse astrocytoma? Fibrillary astrocytoma, as it is the most common histological subtype. Other subtypes recognized by the WHO include gemistocytic and protoplasmic. Diffuse astrocytoma is responsible for what percentage of astrocytic brain tumors? 10 to 15% What is the peak age of incidence? 30 to 40 years of age9 Where in the brain is it most commonly found? Supratentorially, in the frontal and temporal lobes What are common presenting signs and symptoms? • Seizures (50%) • Subtle changes in speech, sensation, vision, or motor function • Personality changes (if located in frontal lobe) How does the tumor appear on CT scans? Typically as an ill-defined, homogeneous mass of low density without enhancement On MRI? T1: hypointense T2: hyperintense Does it enhance with gadolinium? No Fig. 35.4 Low-grade astrocytoma. (A) FLAIR T2 demonstrates a hyperintense frontal lesion. (B) No enhancement is seen in a T1-weighted postgadolinium image, characteristic of a low-grade, rather than anaplastic, lesion. Are mitoses present microscopically in fibrillary astrocytoma? No, nor is necrosis or microvascular proliferation present. Increased cellularity and nuclear atypia (enlarged, cigar-shaped, or irregular hyperchromatic nuclei) are diagnostic. Is fibrillary astrocytoma GFAP positive? Yes, and often S-100 positive as well What is the growth fraction by Ki-67/MIB-1 labeling index? Typically <4% What is necessary for a diagnosis of gemistocytic astrocytoma? >20% of all tumor cells are gemistocytes (usually approximately 35%) How do gemistocytes appear? Plump, glassy, eosinophilic cell bodies with stout processes; eccentric nuclei, usually with distinct nucleoli Are they GFAP positive? Yes What tumor markers do they express? p53 and bcl-2 Though the Ki-67/MIB-1 growth fraction is also <4%, what is the difference in prognosis with gemistocytic versus fibrillary astrocytoma? The gemistocytic variety is more prone to progress to anaplastic astrocytoma. What is the genetic susceptibility associated with diffuse astrocytoma? TP53 germline mutations and Li-Fraumeni syndrome What other genetic change is observed in diffuse astrocytoma? Increased mRNA expression of platelet-derived growth factor receptor-α Is there a consistent way to judge time to progression of higher grade tumor? No; early detection of p53 changes has not been shown to correlate. However, IDH1 and IDH2 mutations may predict a less rapid progression. Fig. 35.5 At low magnification, diffuse astrocytomas infiltrate the brain parenchyma, giving it a hypercellular appearance (A). The tumor cells have irregular, elongated or slipper shaped, hyperchromatic nuclei. The presence of mitoses (B, arrow) corresponds to a diagnosis of anaplastic astrocytoma, WHO grade III. The presence of necrosis (often pseudopalisading) (C) and microvascular proliferation (D) is diagnostic of glioblastoma, WHO grade IV. What are clinical predictors of better prognosis? Younger age, smaller tumor, seizure as only presenting syndrome (rather than focal neurological deficit) Which WHO grade II neoplasm is characterized by a “chicken wire” vascular appearance? Oligodendroglioma Elaborate on what “chicken wire” vasculature means. The lesion is characterized by a delicate network of branching capillaries, which may look like chicken wire.10 To what does a “fried egg” appearance refer? Artifactual perinuclear clear space following delayed formalin fixation. (Because of its “chicken wire” vasculature and “fried egg” appearance on permanent sections, it may be helpful to think of oligodendroglioma as the “chicken” tumor.) Will the tumor have a “fried egg” appearance seen in smear preparations or frozen sections? No What about rapidly fixed tissue or permanent sections made from frozen material? The clearing may or may not be present in these samples. Oligodendroglioma is responsible for what percentage of brain tumors? 2.5% of all primary tumors and 5 to 6% of all gliomas11 What is the typical age at presentation? 40 years of age12 How do most patients with the lesion present (clinically)? Two-thirds of patients present with seizures.11,12 True or false: Patients with oligodendrogliomas have an increased risk of hemorrhage. True; their risk is approximately 20%. Is there a certain etiology for this tumor? No. Associations have been suggested with chemical carcinogens, previous radiation, and viral elements, but no conclusive studies have shown a significant relationship. What is its localization? Cortex and white matter of the frontal lobe > temporal lobe > parietal lobe > occipital lobe How does it appear on CT and MRI? CT: hypo- or isodense, well circumscribed, with or without calcification MRI: T1: hypointense, T2: hyperintense What would a heterogeneous appearance on imaging suggest? Intratumoral hemorrhage/cystic degeneration Fig. 35.6 Oligodendroglioma. CT with contrast (A) and T2-weighted MRI (B) show a well-demarcated corticosubcortical lesion in the left parietal lobe with a cystic degeneration component (asterisk) and calcification (arrow). Such extensive edema is typically associated with anaplastic (grade III) oligodendrogliomas. Aside from a “fried egg” appearance, what features would you expect to see on microscopy? • Increased cellularity • Uniform, round to oval nuclei in low-grade oligodendrogliomas • Tumoral cells may be larger, with eccentric cytoplasm (mini-gemistocytes) • Occasional mitoses • Microcalcifications What if there are frequent mitoses? Then the lesion is probably more consistent with an anaplastic oligodendroglioma (WHO grade III) What other microscopic findings would you expect with an anaplastic oligodendroglioma? • Increased nuclear pleomorphism • Prominent microvascular proliferation • Areas of necrosis, including pseudopalisading necrosis Fig. 35.7 At low magnification, oligodendrogliomas are diffusely infiltrative (A). They may show areas with microcystic change (B) and a prominent “chicken-wire” vascular pattern (C). The cells of WHO grade II oligodendrogliomas are relatively uniform, with round to oval nuclei and perinuclear halos (C). Anaplastic oligodendrogliomas (WHO grade III) show more cellular areas, with brisk mitoses and greater cytological atypia (D), as well as microvascular proliferation. Is oligodendroglioma S-100-positive? Yes. It is also positive for HNK1 (antibody to Human lymphocytes with Natural Killer cell) and microtubule associated protein-2 (MAP2), and other nonspecific markers for neuroectodermal tumors. Is oligodendroglioma GFAP-positive? Sometimes (most commonly seen in mini-gemistocytes and gliofibrillary oligodendrocytes) What other lesions might you want to include on the differential with oligodendroglioma? • Pilocytic astrocytoma • Clear cell meningioma • Neurocytoma • Clear cell ependymoma • Dysembryoplastic neuroepithelial tumor (DNT) How can you differentiate these lesions? • Pilocytic astrocytoma: foci of classic pilocytic features • Clear cell meningioma: periodic acid-Schiff (PAS) positive and epithelial membrane antigen (EMA) positive • Neurocytomas: typically located in the lateral ventricles, have neuropil islands, bear immunoactivity for synaptophysin and NeuN • Clear cell ependymomas: have ependymal and perivascular rosettes, and show focal dot- and ring-like EMA+ • DNTs: may show “floating” neurons, and the neuronal component can be highlighted by neuronal markers What is the most common genetic alteration in oligodendrogliomas? Combined loss of heterozygosity of chromosomes 1p and 19q What does this alteration signify for the patient’s prognosis? Better response to chemotherapy; improved survival rates In a patient with 1p/19q deletion, would you expect to see p53 mutation and/or loss of 17p? No, these alterations are mutually exclusive. (Note: p53 mutations are rare in oligodendrogliomas.) What other genetic alterations may be associated with the lesion? • IDH mutations • Amplification of epidermal growth factor receptor (EGFR) • Loss of chromosome 22 Which genetic changes are associated with anaplastic oligodendroglioma? Losses of chromosomes 9 and 10 What is the prognosis for a patient with oligodendroglioma? 5-year survival: 71% 10-year survival: 54% Median survival: 16.3 years13 Most studies suggest a more favorable prognosis with oligodendroglioma than with diffuse astrocytoma.4 What is an oligoastrocytoma? A diffusely infiltrating glioma that is a mix of cells resembling both oligodendroglioma and diffuse astrocytoma What is its WHO grade? II In what age range and sex would you expect to find this tumor? 35- to 45-year-old man (though there is only a slight male predominance) Is there a virus associated with this lesion? Yes; JC virus sequences have been detected in human oligoastrocytomas, but a definitive link between the two has yet to be found. What is the typical survival time with this tumor? 6.3 years 5-year survival: 58% 10-year survival: 32% Better prognosis associated with loss of 1p/19q (as with pure oligodendroglioma) Which tumors are considered to be high-grade gliomas? • Anaplastic astrocytoma (AA) • Anaplastic oligodendroglioma (AO) • Anaplastic oligoastrocytoma (AOA) • Anaplastic ependymoma • Glioblastoma (GBM) Which tumor variable is most directly related to prognosis? Histopathological grade, but age at presentation and KPS score are also significant. What scale (based on recursive partitioning statistical analysis [RPA]) is widely used to predict survival in patients with high-grade glioma? The Radiation Therapy Oncology Group’s scale, which grades patients from I to VI14 What is the definition of class I, and what is the prognosis with this class? Median survival: 58.6 months 2-year survival: 76% Definition: age <50 years, anaplastic astrocytoma (AA), normal mental status Median survival: 37.4 months 2-year survival: 68% Definition: age ≥50, KPS 70–100, AA, ≥3 months from first symptoms to beginning treatment Class III? Median survival: 17.9 months 2-year survival: 35% Definition: age <50, AA and altered mental status or age <50, GBM, and KPS 90+ Class IV? Median survival: 11.1 months 2-year survival: 15% Definition: age <50, GBM, and KPS <90 or age ≥50, KPS 70–100, AA, and ≤3 months from first symptoms to beginning treatment or age >50, GBM, surgical resection, and good neurological function Class V? Median survival: 8.9 months 2-year survival: 6% Definition: age ≥50, KPS 70–100, GBM, and either surgical resection and impaired neurological function or biopsy only followed by ≥54.4 Gy of radiotherapy or age ≥50, KPS <70, normal mental status Class VI? Median survival: 4.6 months 2-year survival: 4% Definition: age ≥50, KPS <70, abnormal mental status or age ≥50, KPS 70+, GBM, biopsy only, <54.4 Gy of radiotherapy15 Why is surgical resection not indicated when a patient already has significant neurological deficit? Preoperative deficit is the best indicator of postoperative outcome, so patients who are already significantly affected are less likely to have good postoperative course. What is the role of radiotherapy in the treatment of high-grade gliomas? It remains the most effective adjuvant available. How is it administered? 180 cGy fractions to a total dose of approximately 60 Gy to treat the bulk tumor plus the margin of increased T2 signal15 What is the role of chemotherapy as an adjuvant in the treatment of high-grade gliomas? • Response rates are on the order of 30 to 40%. Overall, survival has been shown to increase by a few months.16 • PCV therapy (procarbazine, chloroethylcyclohexylnitrosourea [CCNU], and vincristine) is useful for AO and AOA, especially lesions with deletion of 1p or 1p/19q.15 • Temozolomide is an oral chemotherapeutic agent that has been shown to increase survival when used as an adjuvant in high-grade glioma treatment. • Interferon therapy is currently under trial and has shown promising phase 1 results when combined with temozolomide in the adjuvant treatment of high-grade glioma. What is temozolomide? • Temozolomide is an oral alkylating agent that acts as an MGMT (O6-methylguanine-DNA methyltransferase) inhibitor. • It is FDA approved in initial relapse of anaplastic astrocytoma that had been treated with nitrosourea or de novo GBM. • It is now the standard of care to use this agent as adjuvant in recurrent high-grade gliomas. • Off label use includes newly diagnosed AA, progressive low-grade astrocytomas, and oligoastrocytomas. Can anaplastic astrocytoma arise de novo? Yes, it may progress from a diffuse astrocytoma or arise de novo. What is its WHO grade? III What is its mean age of presentation? Typically mid-40s What tumor does its behavior closely mimic? Diffuse astrocytoma, in terms of localization, clinical signs and symptoms (though it may follow a more rapid course of progression), and histology How does it differ from a diffuse astrocytoma microscopically? Increased cellularity, distinct nuclear atypia (variation in nuclear size and shape and increasing nucleolar prominence), and mitotic activity What Ki-67/MIB-1 growth fraction would you expect with this lesion? 5 to 10% What are the major genetic changes associated with AA? • High frequency of p53 mutation/loss of 17p • Unlike diffuse astrocytoma, loss of 19q What is the prognosis with this lesion? Likely progression to GBM (over an average of 2 years)17 What is the most common primary brain tumor? GBM (meningiomas are considered the most common primary “intracranial” tumor)18 What is its WHO grade? IV Where does it most commonly occur? Cerebral hemispheres: temporal > parietal > frontal > occipital What other sites are common in children? • Basal ganglia and thalamus • Brainstem (“malignant brainstem glioma”) What causes a “butterfly glioma?” Rapid invasion of the lesion through the corpus callosum into the contralateral hemisphere Fig. 35.8 Characteristic “butterfly” appearance of GBM invasion of the corpus callosum on a T1-weighted image with contrast, axial view. Invaded structures serve as a “highway” for the length of their projection, allowing invading cells to exist outside of the contrast-enhancing rim of the tumor, leading to increased risk of recurrence after resection and radiotherapy. True or false: GBM often invades the subarachnoid space and metastasizes via CSF. False, though peritoneal metastasis via VP shunt has been seen, invasion of the dura, bone, vessel lumen, and venous sinus are all exceptional. How do the cysts of GBM differ from cysts seen in low-grade astrocytoma? They are areas of liquefactive tumor necrosis as opposed to retention cysts. Do primary or secondary GBM arise more commonly in younger patients? Mean age for secondary (having progressed from a lower grade astrocytoma) GBM is 45, approximately 10 years younger than for primary GBM. How is this significant for prognosis? Younger patients have a better life expectancy; it is not fully understood if age alone or the secondary nature of these tumors confers a survival advantage (7.8 months vs. 4.5 months).17 Are primary or secondary GBMs more common? Primary (approximately 90%) Aside from differences in prognosis, why is it important to differentiate between primary and secondary lesions? With the exception of loss of heterozygosity of 10q, the lesions have different genetic profiles and they differ in response to therapy. What are essential histopathological findings in GBM? • Anaplastic glial cells • Microvascular proliferation • Necrosis What are “secondary structures?” Morphological findings due to interaction between glioma cells and host brain structures (e.g., subependymal region), around neurons and blood vessels, aka “satellitosis,” or in the subpial zone of the cortex. They exemplify the infiltrative nature of gliomas. What is the typical proliferative index associated with GBM? 15 to 20% by Ki-67/MIB-1 labeling True or false: Glioblastomas are highly vascularized. True What are the three mechanisms of GBM vascularization? • Vessel co-option • Classic angiogenesis • Vasculogenesis (homing of blood marrow cells that encourage growth) What is the major driving force of GBM angiogenesis? Hypoxia, via intracellular stabilization of the hypoxia regulating gene hypoxia-inducible factor 1? (HIF-1?), which activates >100 genes that control angiogenesis. What is the mechanism of vascular dysfunction in GBM? Vascular endothelial growth factor (VEGF) is produced by hypoxic perinecrotic palisading cells to increase angiogenesis, vascular permeability (edema), and homing of bone marrow–derived cells. What is the hallmark feature of necrosis in GBM? Pseudopalisading pattern, which is equally common in primary and secondary lesions. These cells have high rates of apoptosis, low rates of proliferation, and strong expression of HIF-1? and VEGF. True or false: Of all astrocytomas, GBM is associated with the greatest number of genetic changes. True What are the major genetic changes associated with GBM? • Loss of heterozygocity (LOH) of chromosomes 10q (63–70% of first-degree and second-degree GBM) and 17p • EGFR (epidermal growth factor receptor) amplification/overexpression Alteration of p53 is associated more with primary or secondary GBM? Secondary GBM (>65%). It is almost always present in precursor lesions as well. Only about a quarter of primary lesions have associated p53 mutations. Are there any other genetic changes that present more commonly in secondary than in primary GBM? • IDH mutations • LOH 19q (54% vs. 6%) • LOH 22q (82% vs. 41%) Are EGFR changes present in young patients with GBM? No, there have been no reported cases of anyone <35 years of age with EGFR changes in GBM. The amplification is more associated with primary disease (36% vs. 8% in secondary).4 What other genetic changes are more commonly associated with primary than secondary GBM? • Mutation of PTEN, a tumor suppressor gene (25% vs. 4%) • Deletion of p16INK4a, a negative inhibitor of mitosis progression from G1 to S phase (31% vs. 19%) What is the genetic profile of pediatric high-grade astrocytoma? Typically de novo development: • High frequency of p53 mutations (approximately 40%) in older children • Low frequency of p53 mutations in children <3 (12%) • Absence of IDH mutations • Low frequency of EGFR amplification (6%), p16INK4a deletion (19%)4 What variant of glioblastoma is associated with de novo presentation in patients with mean age 42? Giant cell glioblastoma What are the special features of this variant? • Bizarre, multinucleated giant cells • Often abundant reticulin network • Frequent p53 mutations and PTEN mutations Fig. 35.9 GBM variants. (A) Giant cell glioblastoma can be distinguished from PXA by the presence of frequent mitoses, microvascular proliferation and tumor necrosis. (B) Gliosarcoma is composed of malignant spindle cells admixed with conventional GBM. (C) Silver stain highlights pericellular reticulin deposition in the spindle cell areas, and is negative in the area of conventional GBM. (D) Immunostain for GFAP, in contrast, is positive in the areas of conventional GBM and is negative in the spindle cells. Which variant has a genetic and population profile that is very similar to primary GBM, yet is characterized by a “marbled” pattern of glial and mesenchymal differentiation? Gliosarcoma What is this variant’s defining characteristic? GFAP-negative, clearly malignant mesenchymal component, with abundant pericellular reticulin deposition. The malignant astrocytic component is GFAP-positive. What characterizes gliofibroma? Gliofibroma is a variant that mostly affects children (mean age = 14 years) in the cerebrum or spinal cord comprising a glial component and a nonsarcomatous fibroblastic component. This lesion often lacks necrosis and vascular microproliferation. It often follows an indolent course and thus has a much more favorable outcome than GBM Are there any familial syndromes associated with GBM? • Turcot syndrome • Li-Fraumeni syndrome • NF1 • Multiple enchondromatosis What are some of the characteristics of GBM that make it so difficult to treat? • Poor delivery of chemotherapeutic agents due to the blood–brain barrier • Genomic instability resulting in heterozygous cell population that is difficult to treat with a single agent • Invasiveness of the tumor (ability to cross midline and intact blood–brain barrier) • Neural-stem-cell–like cells that may harbor resistance mechanisms • Retention of DNA repair machinery that makes chemotherapy and radiotherapy less effective What method of drug delivery is increasingly being optimized for GBM treatment? Convection-enhanced delivery How does it work? Intraparenchymal catheters are implanted in the tumor bed and attached to a pump that provides constant, positive pressure for the infused chemotherapy agents. What is a major problem with the technique, and how is it being ameliorated? Reflux along the catheter track: • Step-design catheters have a thicker (0.2 mm) guide needle with thinner (0.168 mm) tubing that extends several millimeters beyond the guide’s end. • Increasing the volume of distribution with multi-tipped or hollow fiber catheters • Balloon-tipped catheters inflate to create intraparenchymal space where the therapy can be delivered.19 What is a major bias affecting most trials of GBM therapy? Patients selected for novel treatment studies tend to be younger (<50) and have higher KPS scores, factors that improve their prognosis already. What is gliomatosis cerebri (GC)? Involvement of at least three cerebral lobes (usually bilateral) by diffuse glioma (usually astrocytic) with frequent extension to the brainstem, cerebellum, and spinal cord What is the WHO grade of this lesion? Its biological behavior corresponds to WHO grade III Which MRI sequences are particularly useful in evaluating GC? FLAIR and T2-weighted sequences What is the difference between primary and secondary GC? Primary: extensive CNS involvement at initial presentation Secondary: progressive extension observed over follow-up of initial local diffuse glioma How does GC differ from leptomeningeal gliomatosis? Leptomeningeal gliomatosis is the widespread infiltration of the subarachnoid space by a diffuse glioma. How does GC differ from gliomatosis peritonei? Gliomatosis peritonei results from dissemination of glial tissue throughout the peritoneum, usually in association with ovarian teratoma. What are the microscopic features of GC? They correspond to the type and grade of the diffuse glioma present. Aside from astrocytoma, what other types of tumor may present with a GC pattern? Oligodendroglioma and oligoastrocytoma What genetic alterations have been described in gliomatosis cerebri? TP53 mutations have been identified, although with a lower frequency than diffuse astrocytoma. IDH1 mutations have been identified in type 2 GC, but not in “classic” type 1 GC. 1p deletions have also been identified in cases with oligodendroglial morphology. However, combined 1p/19q loss is not found.20 What is the prognosis with GC? Like all gliomas, it depends on the histological subtype of cells present, the patient’s age, and the KPS score (e.g., an oligodendroglial GC with 1p deletion in a 35-year-old with KPS 90 would carry a more favorable prognosis than an astrocytic GC with multiple mitoses in a 70-year-old with KPS 60). What kind of intracranial neoplasm is most common when autopsy data are considered together with tumors of clinical significance? Meningioma. Incidental meningiomas are found at autopsy in 3% of those over 60 (in 2.3% of autopsy patients overall).21 What is a meningioma? A neoplasm arising from arachnoid cap cells (specialized cells of arachnoid granulations) within the leptomeninges What is its WHO grade? About 94% are WHO grade I, but there are atypical (approximately 5%) meningiomas corresponding to WHO grade II and anaplastic meningiomas (approximately 1%) that correspond to WHO grade III.22 Where in the CNS does it most commonly localize? • Cerebral convexities (especially parasagittal, in association with the falx and venous sinus) (44%) • Sphenoid ridges (11.9%) • Olfactory grooves (9.8%) • Optic nerve sheath • Para/suprasellar regions • Petrous ridges • Posterior fossa • Thoracic spine23 How does its incidence relate to age? Increases with age; most common in middle-aged and elderly, peaking in the sixth and seventh decades Does it preferentially affect one sex? Yes, women are more affected than men, particularly spinal meningiomas. However, atypical meningiomas are more prevalent in men.4 Can meningioma occur in children? Yes, though rarely. Pediatric meningiomas are more likely to be intraventricular and aggressive than are adult lesions.1 Meningiomas associated with hereditary tumor syndromes (e.g., neurofibromatosis) also occur in younger patients. Is trauma a cause of meningioma? Probably not. Though anecdotal evidence suggests a higher incidence of meningiomas in trauma patients, a systemized review found that this phenomenon is probably due to a higher rate of detection of asymptomatic “incidentalomas.”24 What about radiation? Yes. Both low- and high-dose radiation have been linked to meningiomas (latency to tumor ranges from 19 to 35 years, inversely related to the dose of radiation). Radiation-induced meningiomas more commonly occur among younger patients, are multifocal, and are higher grade. Is it common for patients to have multiple meningiomas? No; less than 10% of meningioma patients exhibit multiple lesions. The incidence of multiple lesions is higher in patients with familial syndromes (NF-2)1 and secondary to high-dose irradiation. Why is it difficult to understand the “natural history” of meningiomas? Because historically only those that were symptomatic were detected, whereas in the past 20 years, imaging studies have increased the finding of asymptomatic “incidentalomas” So what is a useful way to estimate the “natural history” of these lesions? Measuring the doubling time of residual tumor volume (Td) after resection, with or without bromodeoxyuridine (BUdR) or Ki-67 labeling. Magnetic resonance spectroscopy (MRS) is also being investigated as a potential noninvasive means of assessment. What is the imaging modality of choice to examine meningiomas? MRI, with and without gadolinium, to determine the full extent of lesion and associated edema. T1: most lesions are isointense (or slightly hyperintense) With contrast: dramatic enhancement, especially of the “dural tail”25 T2: hyperintense or isointense Fig. 35.10 Axial T1-weighted postgadolinium image revealing a convexity meningioma in the frontotemporal region with a visible dural tail (arrow). What is an advantage of CT versus MRI? CT is better for demonstrating calcification within the lesion as well as hyperostosis in the adjacent part of the skull. What are the three most common histological subtypes of meningioma? • Meningothelial • Fibrous • Transitional How would you characterize a meningothelial meningoma? Lobules of tumor cells may be demarcated by thin collagenous septa (large lobules should not be confused with the “sheeting” of atypical meningioma). Whorls and psammoma bodies may be present, but tend to be poorly formed. Groups of uniform epithelioid cells appear syncytial, with indistinct cell borders. Nuclei have delicate chromatin and occasional central clearing or intranuclear cytoplasmic pseudoinclusions. What pattern is characteristic of a fibrous meningioma? Spindle-shaped cells in a collagen-rich matrix with infrequent whorls, psammoma bodies. Focal groups with meningothelial-type nuclei are present. What pattern is characteristic of a transitional meningioma? Tight whorls, psammoma bodies, and a coexistence of meningothelial and fibrous patterns What is a psammomatous meningioma, and where would you expect to find it? A meningioma with more psammoma bodies than tumor cells. These may become confluent, forming irregular calcified masses or bone. They are commonly found in the thoracic spinal region of middle-aged women. Describe an angiomatous meningioma. These lesions feature more blood vessels than tumor cells. Most of the vessels are small with markedly hyalinized walls. Degenerative nuclear atypia is seen commonly. Though not an aggressive lesion, adjacent cerebral edema may be marked. What is a secretory meningioma? Pseudopsammoma bodies (lobules containing PAS-positive, eosinophilic secretion within intracellular lumina. These cells are immunoreactive for CEA (carcinoembryonic antigen). What is the most common genetic abnormality in meningiomas? Deletion of band q12 on chromosome 22, which occurs in approximately half of the cases Which three meningioma subtypes are considered WHO grade II lesions? • Clear cell meningioma • Chordoid meningioma • Atypical meningioma Describe clear cell meningioma. Composed of clear, polygonal cells with glycogen-rich cytoplasm and prominent collagen. This variant is PAS-positive and has diastase-sensitive cytoplasmic clearing because of its glycogen accumulation. It is found in the cerebellopontine angle and cauda equina region, frequently in children and young adults. It frequently recurs, and may have CSF seeding. What is chordoid meningioma? A tumor resembling chordoma, with cords of eosinophilic, vacuolated cells in an abundant mucoid matrix background. These groups are frequently interspersed with more typical meningioma. It is typically a large, supratentorial tumor that recurs frequently after subtotal resection. What characterizes atypical meningioma? Increased mitotic activity (4+ mitoses/10 high-power fields) and/or brain invasion, and/or three or more of the following features: increased cellularity, high nuclear/cytoplasmic ratio, prominent nucleoli, patternless/sheet-like growth pattern, and foci of necrosis. Fig. 35.11 Whorls can be readily identified on cytological preparations of meningioma (A). Meningothelial cells are arranged in cohesive syncytium-like groups, with indistinct cytoplasmic borders, uniform round-to-oval nuclei with fine chromatin, and inconspicuous nucleoli (B). Atypical meningioma, WHO grade II, show four or more mitoses per 10 high-power fields (C, arrows). Areas of geographic necrosis may be also seen (D).
Brain Tumors
35.1 Primary Tumors
35.1.1 Low-Grade Gliomas
35.1.1.1 Basic Concepts
35.1.1.2 Low-Grade Astrocytoma
35.1.1.3 Oligodendroglioma
35.1.1.4 Oligoastrocytoma
35.1.2 High-Grade Glioma
35.1.2.1 Basic Concepts
35.1.2.2 Anaplastic Astrocytoma
35.1.2.3 Glioblastoma (GBM)
35.1.2.4 Gliomatosis Cerebri
35.1.3 Meningiomas
Related posts:
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree



Brain Tumors
Only gold members can continue reading. Log In or Register to continue



