▪ INTRODUCTION
On August 1, 1966, Charles Joseph Whitman, a student at the University of Texas in Austin, murdered his wife and mother before ascending to the observation deck of his school’s 27-story administration building. Having been trained as a sniper in the Marines, he embarked on a shooting rampage, killing 14 and wounding 31 others before being fatally shot by police. His suicide note requested an autopsy, which revealed glioblastoma in the hypothalamus. Mr. Whitman had seen a psychiatrist several months earlier and he had complained of intense anger and described thoughts about shooting people.
Although this is an exceptionally tragic event involving a brain tumor, it does illustrate the need for psychiatrists to be familiar with neurological conditions that could affect personality and cognition. This chapter provides an overview of those aspects of brain tumors that are frequently tested during in-service and board examinations.
The neurological effects of a brain tumor depend on its location. Brain tumors are divided into those that arise from structures of the central nervous system (CNS) (primary brain tumors), and those that are metastatic to the brain.
▪ CLINICAL FEATURES OF BRAIN TUMORS
The most common presenting symptoms in patients with brain tumors include headaches, seizures, cognitive impairment, personality changes, and focal neurological deficits. The location of the tumor and its histology influence the presentation. Additionally, in neurological diagnosis, the time course is extremely helpful in determining the nature of a lesion. A slow-growing mass lesion is often responsible for subacute cerebral dysfunction, particularly in older individuals.
Headache is a presenting symptom in approximately 35% of newly diagnosed brain tumors. During the course of the disease, headache is reported in up to 70% of patients. It is often dull, nonthrobbing, and intermittent. Supratentorial masses can result in frontal headache. Posterior fossa masses cause headache in occipital and cervical areas. Early morning headache due to increased intracranial pressure (ICP) in the recumbent position, is considered a classic presentation. However, this classic presentation occurs in a minority of patients. When a headache is unilateral or worse on one side, this frequently indicates the side of the lesion. A unilateral or bilateral frontal localization is most common for supratentorial tumors, because most pain-sensitive structures (e.g., large blood vessels and meninges) in the cranium are innervated by the trigeminal nerve. Headache as the sole manifestation of an underlying brain tumor is rare, but certain “red flag” features should raise suspicion for a mass lesion. These are intuitive and include new onset of headache, particularly in older patients; a significant change in character from previous headaches; progressive worsening over days or weeks; pain that is present on awakening or is severe enough to cause awakening; worsening with bending over, coughing, or
sneezing; the presence of neurological signs or symptoms; and the presence of nausea, vomiting, or papilledema.
Seizures are the presenting feature of brain tumors in approximately one third of cases. Low-grade tumors, particularly oligodendrogliomas, have a tendency to present with seizures (>70% in some series). Even if seizures do not occur at presentation, they may later occur in 40% to 60% of patients with brain tumors. Seizures tend to be partial, with possible secondary generalization. Seizure semiology may offer clues to the location of the lesion. Seizures involving right gaze deviation and clonic movements of the right arm are likely to originate from the left frontal lobe. Seizures characterized by olfactory hallucinations, déjà vu, impairment of consciousness, and automatisms implicate the temporal lobe. The seizure frequency varies among patients. In patients who have experienced an extensive surgical resection, there may be a marked improvement in seizure frequency (or even complete seizure-freedom) if the active seizure focus has been removed. Conversely, worsening of seizure type or seizure frequency may herald radiological tumor progression.
Altered mental status is a presenting feature in 15% to 20% of cases. Tumors associated with elevated ICP, gliomatosis cerebri, and those located in the frontal lobes are more likely to be associated with altered mental status at presentation. The severity can range from mild inattention to deep coma. Some signs and symptoms also may have localizing value. Frontal lobe tumors can cause progressive change in behavior, mood, and personality, that may be mistakenly attributed to depression; one clue is that these symptoms are relatively resistant to medical therapy.
Focal neurological deficits such as aphasia, hemiparesis, sensory loss, and visual field loss may also occur at presentation and correlate with tumor location. Aphasia is associated with involvement of the dominant hemisphere (usually the left), whereas sensorimotor and visual deficits can occur if the tumor affects the corresponding pathways within the CNS.
Workup of a Patient Suspected of a Brain Tumor
A patient suspected of possibly having a brain tumor should undergo a thorough general and neurological evaluation. Imaging of the brain should be performed in all patients, with magnetic resonance imaging (MRI) preferred over computed tomography (CT) because MRI provides a level of anatomical and pathological detail not possible with CT. Contrast should be given in all patients suspected of having a CNS neoplasm. In certain patients, imaging of the spine may also be considered to exclude drop metastases. The presence of meningeal enhancement on MRI would warrant cerebrospinal fluid (CSF) evaluation to confirm leptomeningeal metastasis. If the patient is suspected of having a metastasis to the brain, a thorough systemic workup for the primary neoplasm should be undertaken. If a diagnosis cannot be established by demonstrating likely metastasis from another primary site of cancer, a brain biopsy or surgical resection may need to be performed.
▪ PRIMARY CENTRAL NERVOUS SYSTEM TUMORS
Primary CNS tumors are classified according to the World Health Organization (WHO) 2007 classification. Tumors are graded by the WHO classification from I to IV, with grade IV being the most aggressive. Approximately 70% of pediatric brain tumors are infratentorial, and medulloblastoma is the most commonly diagnosed brain tumor in children under the age of 10. Conversely, 70% of adult brain tumors are supratentorial, and the most common histological categories include meningiomas and high-grade astrocytomas. A summary of the WHO classification is provided in
Table 27.1, with notable clinical, radiological, pathological, and molecular features described in
Table 27.2.
Astrocytic Tumors
Astrocytomas are neuroepithelial neoplastic counterparts of the glial cell known as the astrocyte. Their astrocytic lineage is confirmed by positive glial fibrillary acidic protein (GFAP) staining on immunohistochemistry. They can be divided into well-circumscribed and diffuse (infiltrating) categories. Well-circumscribed astrocytomas include pilocytic astrocytoma, pleomorphic xanthoastrocytoma, and subependymal giant cell astrocytoma (all WHO grade I). Infiltrating astrocytomas include diffuse (low-grade) astrocytoma (grade II), anaplastic astrocytoma (grade III), and glioblastoma (grade IV). The latter three neoplasms form an interrelated group, sharing molecular and genetic abnormalities. Over time, diffuse astrocytomas accumulate genetic mutations and can progress to higher grade astrocytomas. The average age at diagnosis increases with increasing grade, with the median age of diagnosis being 34, 41, and 55 years for diffuse astrocytoma, anaplastic astrocytoma, and glioblastoma, respectively.
Pilocytic Astrocytoma
Also known as juvenile pilocytic astrocytoma, pilocytic astrocytomas (PAs) (WHO grade I) tend to occur in the first two decades of life and present as a cyst with an enhancing nodule on MRI. The differential diagnosis for this finding is shown in
Table 27.3. Although they can occur throughout the neuraxis, they tend to arise infratentorially in children. They are the most common gliomas in this age group. They are well-circumscribed, slow growing, and characterized by Rosenthal fibers and eosinophilic granular bodies on pathological examination. Surgical resection is usually curative of PA, as well as several other tumors (
Table 27.4). Malignant transformation, although possible, is rare.
Subependymal Giant Cell Astrocytoma
Subependymal giant cell astrocytomas (SEGAs) are associated with tuberous sclerosis (TS), with approximately 10% of patients with TS developing SEGAs at the foramen of Monro by age 20. A typical presentation is hydrocephalus. In about half of all patients with TS, there is no family history, because the TS genes have a high rate of spontaneous mutation.
Pleomorphic Xanthoastrocytoma
Pleomorphic xanthoastrocytomas (PXAs) (WHO grade II) are large cortical tumors with a predilection for the superficial temporal lobes and often present as seizures. The mean age of diagnosis is 14. Brain imaging may reveal a cystic tumor with an enhancing mural nodule. As the name implies, pathology shows pleomorphism, cellular atypia, and lipid-laden multinucleated giant cells. There is GFAP and synaptophysin positivity. Gross total resection may be curative.
Diffuse (Low-Grade) Astrocytoma
As their name implies, diffuse astrocytomas are quite infiltrative and do not have distinct boundaries with normal brain. Based on the prevailing cell type, three major variants can be distinguished. These are fibrillary (most common), gemistocytic, and protoplasmic. The gemistocytic variant carries a worse prognosis and increased tendency for malignant transformation. Unlike circumscribed astrocytomas, diffuse astrocytomas are not curable with surgical resection. However, there are data to suggest that extensive surgical resection may prolong survival.
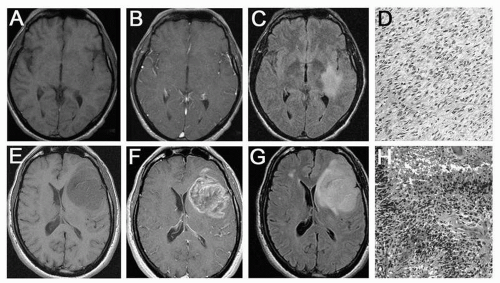
The average age at diagnosis of diffuse astrocytoma is 34 years. The clinical presentation may include seizures and headaches. The tumor may also be discovered incidentally on brain imaging obtained for other reasons, such as trauma. Brain CT reveals a nonenhancing hypodensity, and MRI typically shows a diffuse, nonenhancing area of T2 hyperintensity (
Fig. 27.1A-D). Cyst formation is possible, but calcifications are much less common than oligodendrogliomas. Histologically, the tumor resembles normal brain, but is distinguished by increased cellularity. There is an absence of high-grade features such as high mitotic activity, endovascular proliferation, or necrosis. Mutations of
TP53 (a tumor suppressor gene) can be found.
A needle or open biopsy may be sufficient to establish a diagnosis; however, extensive surgical resection is preferable when feasible. There are two reasons for this. First, an extensive resection yields more tumor tissue to ensure an accurate pathological diagnosis. An erroneous diagnosis of a grade II astrocytoma (when the tumor is actually higher grade) may result in undertreatment and undersurveillance of the tumor. Second, data suggest that an extensive surgical resection of low-grade astrocytomas may impart a survival advantage.
Additional treatment options for low-grade astrocytomas include radiation therapy and chemotherapy, although there is no class I evidence proving a survival advantage with these modalities for low-grade gliomas. The median survival in patients with diffuse astrocytomas is 6 to 8 years, with marked variations between individuals. Because of this relatively longer survival, there are concerns of long-term cognitive toxicity related to therapy (particularly radiation) in patients with low-grade tumors. After a variable period of slow tumor growth, diffuse astrocytomas may transform to a higher grade astrocytoma.
Anaplastic Astrocytoma
Anaplastic astrocytoma (WHO grade III) is a more aggressive malignancy than low-grade diffuse astrocytoma. The median age of diagnosis is older at 41 years. It is histologically characterized by high cellular density and evidence of increased proliferation (mitotic figures). On MRI, the tumor may be associated with more mass effect and may be enhancing. Median survival is 3 to 5 years. After surgery, patients are usually treated with radiation therapy and may receive chemotherapy.