▪ INTRODUCTION
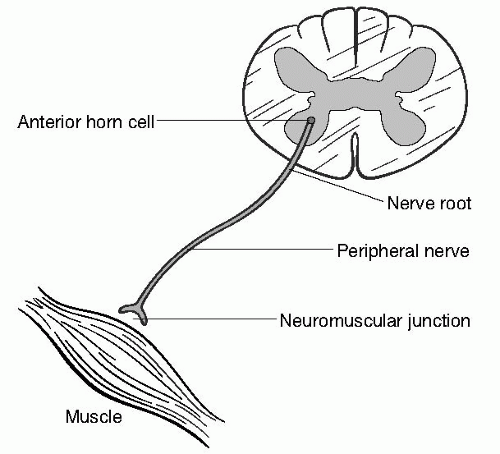
The evaluation of a suspected neuromuscular disorder begins, like the evaluation of all other neurological disorders, with an attempt to localize the problem to a specific level of the neuraxis. The peripheral nervous system encompasses a variety of structures (
Fig. 17.1), and dysfunction at each level produces a characteristic pattern of neurological signs and symptoms.
Motor Neuron and Motor Unit
A motor neuron is a nerve cell with its cell body in the anterior horn of the spinal cord. A motor unit consists of a motor neuron and all the muscles it innervates. The features that suggest a motor neuron disorder include painless weakness, muscle cramps, fasciculations, and muscle atrophy.
Nerve Root
Sensory fibers from the peripheral nervous system have their cell bodies in dorsal root ganglia and enter the spinal cord as dorsal nerve roots. The sensory distribution supplied by a sensory nerve root is known as a dermatome. Motor fibers emerge from anterior horn cells and exit from the spinal cord as ventral nerve roots. The motor territory supplied by a motor nerve is called a myotome. In clinical neurology, the structure that is referred to as the “nerve root” is actually a mixed spinal nerve consisting of the fused dorsal and ventral nerve roots. Characteristics of nerve root dysfunction, or radiculopathy, include pain, paresthesias, and numbness radiating in a dermatomal distribution and weakness in a myotomal distribution.
Plexus
The most anatomically complex structures of the peripheral nervous system are the collections of peripheral nerves known as the brachial and lumbosacral plexuses. The brachial plexus provides sensory and motor innervation to the upper extremity, and the lumbosacral plexus provides sensory and motor innervation to the lower extremity. The identification of plexus disorders can be challenging and relies on demonstrating sensory and motor deficits in the distribution of multiple nerves and nerve roots.
Nerve
Characteristics of nerve dysfunction include pain, sensory loss, paresthesias, and weakness in the distribution of a nerve or multiple nerves. Either sensory or motor findings may predominate, or a mixed sensorimotor picture may be observed. Mononeuropathy involves a single named nerve, mononeuropathy multiplex involves multiple nerves, and polyneuropathy is a generalized process involving all nerves, often (but with many exceptions) with a length-dependent distribution. Axonal neuropathies involve injury to the nerve cell itself, and
demyelinating neuropathies attack the myelin coating of the nerve. This distinction may not be apparent clinically, but axonal and demyelinating neuropathies can be differentiated electrophysiologically, as discussed in the section on neurophysiological testing.
Neuromuscular Junction
The neuromuscular junction is the interface between the nerve and muscle. The nerve releases vesicles containing the neurotransmitter acetylcholine, which binds to postsynaptic acetylcholine receptors on the muscle, resulting in the firing of miniature end-plate potentials. Disorders of the neuromuscular junction produce painless, often fatigable weakness without sensory loss.
Muscle
Muscle disorders produce wasting and weakness without sensory abnormalities, although cramps and myalgias may occur. Most muscle disorders preferentially affect proximal muscles such as neck flexors and extensors, deltoids, iliopsoas, and quadriceps. Distal muscles, extraocular muscles, and respiratory muscles may be involved, however, in specific types of myopathies.
▪ NEUROPHYSIOLOGICAL TESTING
After a careful history and physical examination, further diagnostic testing to establish the localization of a suspected peripheral nervous system disorder may not be necessary. If the localization is in doubt, however, electromyography (EMG) and nerve conduction studies (NCS) may be required.
Nerve Conduction Studies
In NCS, nerves are stimulated with surface electrodes and signals produced by the nerves or the muscles that they innervate are recorded. The most commonly measured NCS parameters are the compound muscle action potential (CMAP) amplitude, sensory nerve action potential (SNAP) amplitude, and conduction velocity.
Late Responses
Late responses are used to assess proximal nerve segments and are recorded using the NCS technique. There are two types of late responses. F waves are produced by stimulation of a motor nerve, conduction of the electrical impulse back toward the spinal cord, firing of a select portion of anterior horn cells, and conduction of the signal back toward the muscle. H reflexes are most commonly obtained by stimulating the sensory component of the tibial nerve, conduction of this signal back to the spinal cord, synapse with the anterior horn cell, and conduction back down the motor nerve to cause contraction of the soleus. The H reflex is the neurophysiological equivalent of the ankle jerk, and it should be emphasized that the stimulus is conducted by both sensory and motor nerves, unlike F waves, which are conducted entirely by motor nerves.
Needle Electromyography
During EMG, a small electrode embedded in a needle is inserted into a muscle, and muscle electrical potentials are analyzed semi-quantitatively. Spontaneous muscle activity is measured by inserting the needle into the muscle while the patient is completely relaxed. Abnormal spontaneous muscle activity (fibrillations, positive sharp waves, myotonia, fasciculations) can be seen in a variety of neuromuscular pathologies. Motor unit activity is recorded and analyzed by having the patient contract the muscle. In chronic neurogenic lesions (anterior horn cell, nerve root, plexus, and nerve), the characteristic EMG findings are long duration, large amplitude, and polyphasic motor units, reflecting reinnervation of denervated muscles. Myopathic motor units are of short duration, of small amplitude, and polyphasic.
Two important EMG concepts are those of activation and recruitment. There are two ways to increase muscular force. The first is by activating an individual motor unit to increase its firing rate, a process mediated by the central nervous system (CNS). The second way to increase muscular force is by recruiting additional motor units, a process mediated by the peripheral nervous system. In CNS disorders, therefore, activation is poor and recruitment is normal. Nerve root and peripheral nerve disorders produce normal muscle activation but decreased recruitment. In combined disorders of the central and peripheral nervous systems (such as amyotrophic lateral sclerosis), both activation and recruitment are abnormal. Muscle disorders are characterized by normal muscle activation and early recruitment.
A brief summary of the expected findings of EMG and NCS is presented in
Tables 17.1 and
17.2.
▪ MOTOR NEURON DISEASES
Amyotrophic Lateral Sclerosis
Amyotrophic lateral sclerosis (ALS) is a progressive and uniformly fatal disorder characterized by wasting and weakness that often begins asymmetrically in an arm or leg. Later, involvement of the other limbs, bulbar muscles, and respiratory muscles occurs. A diagnosis of probable ALS is made when muscles of at least three of four different myotomal levels (bulbar, cervical, thoracic, and lumbosacral) are affected. A combination of upper (spasticity, hyperreflexia) and lower (wasting, weakness, fasciculations) motor neuron dysfunction is required to make a diagnosis, and EMG has a confirmatory role. Familial ALS accounts for only 10% of all cases and is associated with mutations in the superoxide dismutase (SOD) gene in 20% of these cases. Variants of ALS include primary lateral sclerosis (PLS), which involves isolated degeneration of upper motor neurons, and progressive muscular atrophy (PMA), which involves lower motor neurons exclusively. Both of these variants have better prognoses than ALS. Treatment of ALS is largely supportive and includes assistive devices, positive pressure ventilation at night, and the glutamate antagonist riluzole. Despite these treatments, the mean survival is approximately 3 to 4 years, with a universally fatal outcome in those patients who are not mechanically ventilated.
Kennedy’s Disease
Kennedy’s disease is an X-linked bulbospinal neuronopathy caused by a CAG trinucleotide repeat expansion in the androgen receptor gene. The disorder affects men exclusively and is characterized by muscle wasting and weakness, without signs of upper motor neuron dysfunction such as spasticity or hyperreflexia. Fasciculations in the lower face and gynecomastia are often prominent. No specific treatment is available for Kennedy’s disease, although the prognosis is better than that for ALS.
Spinal Muscular Atrophy
The three most common types of spinal muscular atrophy (SMA) are caused by deletions in the survival motor neuron gene on chromosome 5 and are inherited in autosomal recessive fashion.
SMA I (Werdnig-Hoffmann disease) begins between birth and 6 months of age, with death usually occurring before age 2. Presenting symptoms include hypotonia, a weak cry, poor suck, and respiratory distress.
SMA II begins between 6 and 18 months, although symptoms may be present at birth. Delayed motor milestones bring these patients to neurological attention. Most children with
SMA II are able to roll over and sit, but do not achieve the ability to walk independently. Death from respiratory failure occurs in childhood or early adulthood.
SMA III (Kugelberg-Welander disease) may not be evident until after age 5. Some patients with SMA III may remain ambulatory well into adulthood, but most require a wheelchair by the time they are in their mid 30s. Treatment of SMA III is supportive, with the goals being maintenance of ambulation, respiratory support, and prevention of contractures.
Polio
Caused by poliovirus infection, acute poliomyelitis is now uncommon in the industrialized world as a result of childhood immunization. Most patients infected by the virus develop a gastrointestinal illness or are asymptomatic. Acute poliomyelitis, however, begins as aseptic meningitis, followed by flaccid, asymmetrical paralysis developing within a week. Spinal fluid analysis shows polymorphonuclear cells in the acute stage. Treatment is supportive. If bulbar and respiratory muscles are involved, intubation may be required. The post-polio syndrome is characterized by progressive wasting, weakness, cramps, fasciculation, pain, and fatigue many years after an episode of acute poliomyelitis. Supportive care with an emphasis on physical therapy is the mainstay of post-polio syndrome treatment.
Benign Fasciculations (Cramp-Fasciculation Syndrome)
Fasciculations are spontaneous discharges of individual motor units. Although frequently prompting investigation for ALS, fasciculations may also be a benign phenomenon as in cramp-fasciculation syndrome. Benign fasciculations can be distinguished from ALS-associated fasciculations by the absence of associated weakness and denervation changes on EMG. Reassurance that the patient does not have ALS is often helpful in relieving symptoms, although recalcitrant fasciculations may require treatment with agents such as quinine or carbamazepine.
▪ DISORDERS OF THE BRACHIAL PLEXUS
Traumatic Injury
In adults the most common forms of traumatic injury to the brachial plexus are falls and motor vehicle accidents. Violent downward movement of the shoulder results in an upper trunk disorder affecting predominantly the C5-6 nerve roots (Erb’s palsy). Hyperabduction causes a lower trunk injury affecting predominantly the C8-T1 nerve roots (Klumpke’s palsy). In newborns, Erb’s palsy may occur at delivery, with risk factors for the development of the condition including large gestational weight and small pelvic diameter. Brachial plexus injuries in the newborn may require surgery if improvement in muscle strength is not noted within 3 to 6 months.
Neoplastic-Related Brachial Plexopathy
The two main causes of neoplastic-related brachial plexopathy are direct infiltration of the plexus by tumor and radiation-related injury. Infiltrative lesions are usually derived from tumors of the lung or breast, are associated with a higher incidence of pain, tend to affect the lower plexus, and can produce Horner’s syndrome. Radiation-related plexopathy occurs
months to years after radiation therapy and is generally less painful than plexopathy caused by tumor infiltration. A characteristic EMG finding in patients with radiation-induced brachial plexopathy is myokymia—rhythmic, grouped repetitive discharges of the same motor unit. Radiation therapy is the mainstay of treatment of neoplastic infiltration; the treatment for radiation-related plexopathy is mainly supportive.
Idiopathic Brachial Neuritis
Idiopathic brachial neuritis is known by a variety of names, including Parsonage-Turner syndrome, brachial plexitis, and neuralgic amyotrophy. Typically, a patient who develops idiopathic brachial neuritis has an upper respiratory tract infection several weeks prior and then experiences the sudden onset of pain in the arm and shoulder, and in several days to weeks develops weakness in the affected limb. Recovery is generally spontaneous, but may be prolonged over several months. In cases with poor recovery or severe deficits, corticosteroids or intravenous immunoglobulin (IVIg) may be beneficial.
Neurogenic Thoracic Outlet Syndrome
Neurogenic thoracic outlet syndrome is an uncommon syndrome characterized by a lower trunk brachial plexopathy, predominantly affecting T1 fibers. The causes of neurogenic thoracic outlet syndrome include cervical ribs, fibrous bands, and scalene muscle anomalies. Symptoms include pain in the cervical and suprascapular regions, with pain and weakness in the hand. On physical examination, sensory loss is characteristically confined to the medial hand and forearm and weakness is found in intrinsic hand muscles. The mainstay of treatment is physical therapy, with surgical decompression necessary for refractory cases.
▪ DISORDERS OF THE LUMBOSACRAL PLEXUS
Traumatic Injury
Because of the protected location of the lumbosacral plexus, traumatic injury to this structure is less common than traumatic brachial plexopathy. Traumatic lumbosacral plexopathy may occur in women during childbirth, especially in the presence of fetal-pelvic disproportion.
Mass Lesions
Retroperitoneal hematomas, tumors, and abscesses may produce lumbosacral plexopathy. Anticoagulation, pelvic and abdominal surgery, and femoral catheterization may produce hematomas of the lumbosacral plexus. Evaluation should include computed tomography (CT) or magnetic resonance imaging (MRI) of the pelvis to exclude these lesions. Treatment is directed at the underlying source and may involve surgical intervention and reversal of anticoagulation.
Neoplastic Lumbosacral Plexopathy
Like the analogous syndrome affecting the brachial plexus, direct infiltration of the lumbosacral plexus by tumor produces a syndrome of subacute pain and limb weakness. Radiation-induced lumbosacral plexopathy is typically less painful than infiltrative plexopathy and may be associated with myokymia on EMG. Radiation therapy is the treatment of lumbosacral plexopathy related to tumor infiltration, whereas radiation-related plexopathy is poorly responsive to treatment.
Diabetic Amyotrophy
Diabetic amyotrophy occurs in individuals with type 2 diabetes, often with well-controlled diabetes. The subacute development of hip and leg pain that progresses over several weeks precedes proximal leg weakness, which may be severe. The mechanism of diabetic amyotrophy is
not entirely clear, with vasculitic insults, inflammatory processes, and autoimmune attacks against the lumbosacral plexus or nerve roots implicated. Although many patients recover from the condition without treatment, patients may benefit from a course of IVIg or corticosteroids. Treatment must also incorporate strict glucose control.
Idiopathic Lumbosacral Plexitis
Less common than its brachial plexus counterpart, idiopathic lumbosacral plexitis is characterized by the subacute development of pain followed by weakness in the lower extremity. Unlike idiopathic brachial neuritis, however, the prognosis of idiopathic lumbosacral plexitis is more guarded, with a larger proportion of patients having a prolonged course or poor recovery. Treatment with IVIg or corticosteroids may improve outcome.