Chapter 100 Central Sleep Apnea and Periodic Breathing
Abstract
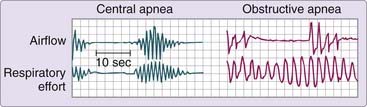
Cessation of breathing during sleep can result from obstruction of the upper airway (obstructive apnea), absence of inspiratory effort (central apnea), or a combination of the two. The term central sleep apnea is used to describe both the pattern of an individual event and the clinical disorder characterized by repeated episodes of apnea during sleep resulting from the temporary suspension of ventilatory effort.1 A central apnea is conventionally defined as a period of at least 10 seconds without airflow due to absence of evident inspiratory effort. This condition differs from the obstructive or mixed apnea by the absence of upper airway obstruction and subsequent inspiratory attempts against the occluded airway (Fig. 100-1). Although this chapter is concerned with central sleep apnea, central and obstructive events are rarely seen in isolation. The vast majority of patients with central apneas also have some obstructive events. This observation suggests that the mechanisms responsible for the different types of apnea must overlap, and research indicates this is likely to be the case.
The muscles of the upper airway (the genioglossus and others) behave as ventilatory muscles,2 dilating or stiffening the pharynx on inspiration. If a decrease or loss of activity occurs in both upper airway muscles and the diaphragm,3 one of several consequences seems likely:
After tracheostomy for obstructive sleep apnea, periodic fluctuations in ventilation with or without central apnea can persist, but they generally resolve over a period of months.4,5 This can also occur when CPAP is initiated to alleviate upper airway obstruction (complex sleep apnea). In addition, it has been observed that the treatment of central apneas with either a respiratory stimulant (e.g., acetazolamide6) or diaphragmatic pacing7 can result in obstructive events. Finally, studies report objective evidence of pharyngeal airway narrowing during purely central apneas.8 These observations imply some commonality in the pathophysiology of the various types of apnea, so it is not surprising that central and obstructive apneas are often seen in the same person.
Patients with predominantly central sleep apnea constitute less than 10% of persons with sleep apnea in most sleep laboratory populations.1,9–11 As a result, only a small number of studies with more than a few such patients have been reported, which makes knowledge of this disorder scant. Most of this chapter provides a discussion of patients with central sleep apnea who breathe normally during the day; however, any patient with hypoventilation during wakefulness almost certainly has hypoventilation with or without central apneas during sleep.
Pathophysiology
Because central apneas are defined as pauses in breathing due to lack of inspiratory effort, a complete loss of electromyographic activity of the ventilatory muscles during such an apnea would be expected, and this has been demonstrated.3 After the apnea, there is a resumption of normal ventilatory muscle activity. This finding implies that the neuronal output to these muscles ceases during central apnea and returns at the end of the ventilatory pause. Central apneas therefore represent a loss of inspiratory drive. Although the cause of central apnea in many patients remains obscure, investigation into the control of breathing has pointed to a number of possible mechanisms.
Control of Breathing
Ventilation is controlled by a number of processes that have been generally grouped under three headings.12 The first is the automatic or metabolic control system, consisting of the chemoreceptors (carotid body for hypoxia and carotid body plus medullary chemoreceptors for hypercapnia), vagally mediated intrapulmonary receptors, and various brainstem mechanisms (namely the pre-Bötzinger complex13) that process the information from these peripheral receptors to generate rhythmic breathing. This metabolic system keeps ventilation regular and ensures that the quantity of ventilation occurring at any time is well matched to metabolic requirements. The second system is the behavioral control system. It seems clear that the activities of normal life, such as talking and eating, can influence ventilation and are thought of as behavioral or volitional influences. The origin of this neural input to respiration is probably in the forebrain. The third ventilatory control process in awake humans and animals is the wakefulness stimulus, with increased ventilation being inherent to the waking state.12 Although the mechanisms responsible for this effect of wakefulness on ventilation are poorly defined, it has been proposed that it either results from the influence of the descending arousal systems on respiratory pattern generators in the brainstem or it is a product of tonic input from nonrespiratory sensory mechanisms such as sight or hearing on the respiratory control system.14,15 The important point is that during wakefulness, ventilation is controlled by both the metabolic and the behavioral systems, including this wakefulness stimulus. Ventilation is likely to persist during wakefulness even with the complete absence of metabolic mechanisms.
During sleep, particularly non–rapid eye movement (NREM) sleep, breathing is controlled almost solely by the metabolic control system, with ventilation being tightly linked to afferent input from chemoreceptors and vagal intrapulmonary receptors.16 This observation was demonstrated in dogs by blocking the input from each of these receptors and monitoring the change in ventilatory pattern. These interventions produced a marked slowing of ventilatory frequency, with long apneic periods. Apnea can also be produced during sleep (with few to no apneas during wakefulness) by partial destruction of the pre-Bötzinger complex in rats.17 The pre-Bötzinger complex is a major site of respiratory rhythm generation and is modulated by chemoreceptor stimuli. In humans, oxygen administration, which reduces the hypoxic stimulus to breathing, has been shown to decrease ventilation during sleep and to initially prolong apneas in some persons in whom such events were already present. In addition, hypocapnic alkalosis, which reduces the hypercapnic ventilatory drive, has been shown to produce central apneas in otherwise normal men.18,19 This combination of studies indicates the importance of both the hypoxic and the hypercapnic influences on breathing during sleep. The implication is that maintenance of neuronal output to the respiratory muscles during sleep may be critically dependent on incoming stimuli, such as those from chemoreceptors.
Considerable investigation has been directed at determining the influence of sleep itself on chemoreceptor activity.20,21 Although this is still controversial, the majority of the available information suggests that ventilatory responses to hypoxia and hypercapnia are reduced somewhat during NREM sleep and fall further during rapid eye movement (REM) sleep.20,21 However, several studies indicate that the decrement during NREM sleep is very likely a product of diminished resistive load compensation (reduced defense of ventilation in the presence of the normal physiologic increase in upper airway resistance associated with NREM sleep) rather than a true loss of chemosensitivity.22 Most investigators believe that chemoresponsiveness is reasonably well maintained during sleep, particularly NREM sleep, and is probably important in maintaining rhythmic ventilation during sleep.
Arterial Carbon Dioxide Tension and Breathing during Sleep
In his classic studies, Bülow23 showed that periodic breathing during sleep was related to PCO2 in an important way. He observed that apnea or pronounced reduction of ventilation occurred “only when the preceding PCO2 was relatively low,” suggesting that a reduced PCO2 during sleep can decrease the drive to breathe to the point of apnea.
Other studies have confirmed the prominent role played by PCO2 in maintaining rhythmic breathing during sleep. Skatrud and Dempsey18,24 showed that passive positive-pressure hyperventilation of sleeping subjects yielded apnea by reducing PCO2 by only 3 to 6 mm Hg below the sleeping value. The actual PCO2 level associated with apnea was often only 1 to 2 mm Hg below the awake value as PCO2 rose from wakefulness to sleep. Each subject had an “apnea threshold,” a PCO2 level below which apnea was commonly seen. It seemed, therefore, that the waking PCO2 level was at or near this apnea threshold, such that waking PCO2 levels may be inadequate to stimulate ventilation during sleep. It was also demonstrated that the periodic breathing during sleep that is often seen with prolonged hypoxia could be abolished by elevating the PCO2 above the predetermined apnea threshold. This finding suggests that the hypocapnia induced by hypoxia, not hypoxia itself, is the pivotal element in this periodic breathing. The authors19 concluded “that effective ventilatory rhythmogenesis in the absence of stimuli associated with wakefulness is critically dependent on chemoreceptor stimulation secondary to PCO2−[H+].”
Subsequent studies by this same investigative group suggest that increased tidal volume (mediated by vagal mechanisms) might also be a primary mechanism inhibiting inspiration after a series of large breaths.25 However, the concept of hyperventilation ultimately leading to an inhibition of inspiratory effort as reflected by a central apnea (whether secondary to hypocapnia or vagal mechanisms), remains intact and is likely to be an important one in understanding the pathogenesis of central sleep apnea.
Ventilatory Control Stability
The mechanisms just described are all seemingly designed to maintain ventilatory stability during wakefulness and sleep, thus avoiding large swings in ventilatory pattern and arterial blood gases. However, abnormalities in the various components of this ventilatory control system can lead to ventilatory instability. This can best be understood by introducing the concept of loop gain, an engineering term that describes the gain of the negative feedback loop that regulates ventilation. Figure 100-2 is a block diagram showing the three major components of this loop: the plant, the circulation delay, and the controller. Plant refers to the lungs, blood, and body tissues where carbon dioxide is stored. Plant gain is the change in alveolar PCO2 for a given change in ventilation. The circulation delay is the time it takes for pulmonary capillary blood to reach the chemoreceptors and is primarily influenced by the cardiac output. The controller represents all the structures responsible for converting chemoreceptor stimuli into ventilation. Controller gain is the change in ventilation for a given change in arterial PCO2.
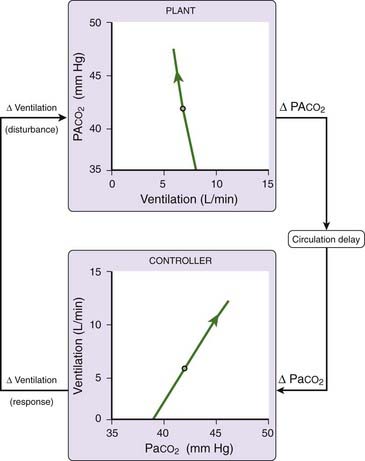
Loop gain, which is the product of the plant and controller gains, is defined as the ventilatory response to a disturbance of ventilation. For example, a reduction in ventilation [D Ventilation (disturbance)], (see Fig. 100-2) produces a change in alveolar PCO2, which, after a delay, produces a change in ventilation [D Ventilation (response)] at the controller. The ratio of the ventilatory response to the ventilatory disturbance is a measure of the stability of the ventilatory control system. A large ratio (high loop gain) indicates an unstable system prone to large swings in blood gases and ventilation. A small ratio, on the other hand, indicates a stable system that exhibits little or no ventilatory fluctuations in response to a disturbance.
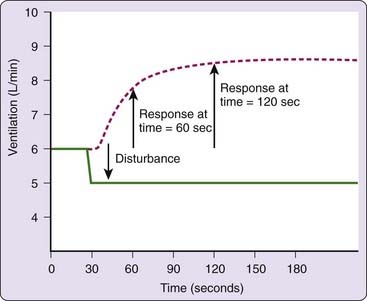
The dynamic aspects of loop gain that are important to ventilatory control stability are illustrated in Figure 100-3. In this figure, the ventilatory control system is disturbed by reducing ventilation slightly and holding it at a reduced level for several minutes. This produces an increase in the ventilatory drive to breathe. Note that it takes time for ventilatory drive (the response) to reach its final, steady-state value and that the ventilatory response to disturbance ratio is maximal when the response achieves a steady state. Thus, loop gain is not a single number, but rather it is a function of (1) the duration of the disturbance and (2) the time it takes for the response to reach a steady state.
Sleep-Onset Central Apneas
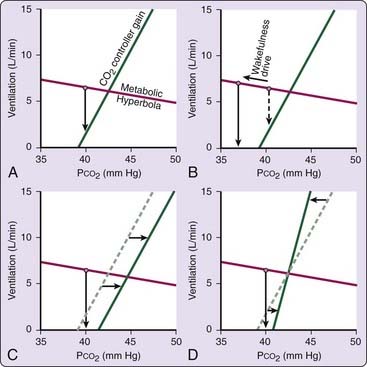
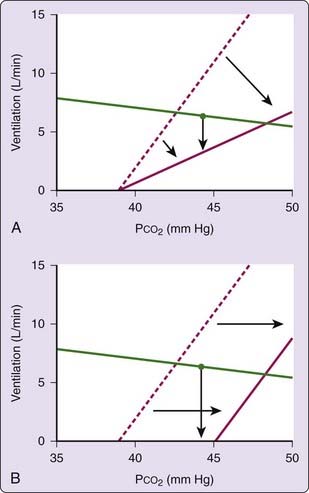
Ventilation is remarkably dependent on the metabolic control system during sleep and the primary stimulus to ventilation during sleep is arterial PCO2. As a consequence, central apneas can occur when the wakefulness drive is lost during the transition from wakefulness to sleep. The mechanism of sleep-onset central apnea is illustrated in Figure 100-4, which shows a plot of the CO2 controller gain during sleep and the metabolic hyperbola. During sleep, the intersection of the two lines marks the steady-state ventilation and PCO2. During wakefulness, however, there is an additional drive to breathe (the wakefulness stimulus), and thus ventilation will be slightly higher (and PCO2 slightly lower) than during sleep. At sleep onset, when the wakefulness drive is withdrawn, central hypopnea or apnea can occur (Fig. 100-4A). A central apnea is likely if there is a large wakefulness drive to breathe (see Fig. 100-4B), if there is a rightward shift in the controller curve (see Fig. 100-4C), or if there is a high controller gain or a flat metabolic hyperbola (see Fig. 100-4D). To the extent that any of these factors are present, an excessive number of central events can occur at the wake-to-sleep transition, producing a pattern indistinguishable from idiopathic central sleep apnea. Likewise, any process that leads to frequent wake–sleep transitions over the course of the night (such as insomnia, obstructive sleep apnea, maladaptation to CPAP, or periodic leg movements) can increase the number of central apneas. It should be clear, however, that nonrepetitive central hypopneas and apneas in the wake-to-sleep transition are probably normal events.
Hypercapnic Respiratory Failure
With loss of wakefulness, ventilation becomes completely dependent on metabolic control mechanisms. In persons with reduced controller gain (i.e., low or absent response to hypoxia or hypercapnia), breathing during wakefulness may be maintained by behavioral or wakeful stimuli. However, during sleep, when these wakefulness mechanisms are no longer operative, there may be little residual drive to breathe, and thus significant hypoventilation can occur. Periodic central apneas, however, would be unexpected due to the low loop gain (Fig. 100-5A). This is generally the case in patients with disorders such as central alveolar hypoventilation (Ondine’s curse)26 and the obesity hypoventilation (pickwickian) syndrome.27 If, however, hypercapnia results from a general reduction in the drive to breathe without significant change in the controller gain—namely, a rightward shift in the controller gain line—then cycling central apneas would be expected (see Fig. 100-5B). This is likely to be the case in patients taking opioid medications, in whom central apneas in association with hypoventilation are common.28,29
Idiopathic Central Sleep Apnea
By definition, the pathogenesis of idiopathic central sleep apnea is not entirely clear. A handful of studies30–33 have shown that these patients tend to have a high hypercapnic response and low arterial PCO2 levels during wakefulness, suggesting that an elevated loop gain may be responsible for the periodic central apneas. However, careful examination of the actual ventilatory pattern in this disorder suggests that other mechanisms might be involved as well. A pure cycling of chemoreceptor (PCO2)-mediated respiratory output would yield a gradual waxing and waning of ventilation, with an apnea or hypopnea at the nadir as is seen in Cheyne-Stokes respiration (see later). In idiopathic central sleep apnea, the ventilatory pauses are often terminated with an abrupt, large breath, not with a gradual increment in ventilation. Although the explanation for this has not been fully elucidated, this pattern strongly suggests that the mechanisms involved in respiratory switching (expiration to inspiration) are affected by this disorder.34 The long expiratory pause that characterizes these central apneas seems to be a failure of the expiratory-to-inspiratory switch, which may be influenced by not only the chemoreceptors but other mechanisms as well (lung volume, chest wall mechanoreceptors, blood pressure). How these inputs individually contribute to this cycling ventilatory pattern is unclear. However, the pattern of central sleep apnea clearly suggests a different mechanism from that of Cheyne-Stokes breathing.
Cheyne-Stokes Respiration
It has long been recognized that congestive heart failure is associated with Cheyne-Stokes respiration during wakefulness and sleep, characterized by a crescendo–decrescendo pattern of tidal volume with a central apnea or hypopnea at the nadir. As stated earlier, this is quite different from the more-abrupt onset and offset of idiopathic central sleep apnea. This breathing pattern is almost entirely a product of ventilatory control system instability (high loop gain) resulting primarily from a prolonged circulation time. As previously discussed (see Ventilatory Control Stability), a prolonged circulation delay means that ventilatory disturbances (e.g., apnea or hyperpnea) go unrecognized, and therefore uncorrected, for a longer time. This leads to greater changes in blood gases and a larger ventilatory response-to-disturbance ratio.
A prolonged circulation delay alone, however, is probably not sufficient in most patients to raise the loop gain high enough to produce Cheyne-Stokes respiration. In anesthetized animals, a several-fold increase in circulation time was needed to induce periodic breathing.35 Such an increase is probably greater than what commonly occurs with heart failure. Thus, an elevated controller gain36 or plant gain are probably also necessary. This might explain why the severity of heart failure does not alone account for the presence or absence of Cheyne-Stokes breathing.
Cheyne-Stokes respiration has been reported in patients with neurologic disease as well, primarily cerebrovascular disorders37; however, the actual ventilatory pattern in these patients has been less well characterized than in patients with congestive heart failure, and the mechanisms remain less well understood.
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


