Chapter 43 Clinical Pharmacology of Other Drugs Used as Hypnotics
Supported by NIH grants MH024652, AG015138, AG020677.
Abstract
Pharmacoepidemiology data indicate that prescribers often use non–benzodiazepine receptor agonist drugs to treat insomnia. Data from the National Disease and Therapeutic Index between 1986 and 1996 show that benzodiazepine hypnotic prescriptions fell by almost 50%, whereas the use of antidepressant drugs to treat insomnia increased by 146%.1 The majority of this increase was accounted for by prescription of trazodone. In fact, trazodone was the most commonly prescribed drug to treat insomnia in 1996. A variety of subsequent data demonstrate that trazodone and other antidepressants continue to be either the first or second most commonly prescribed hypnotic agents in the United States.2–4
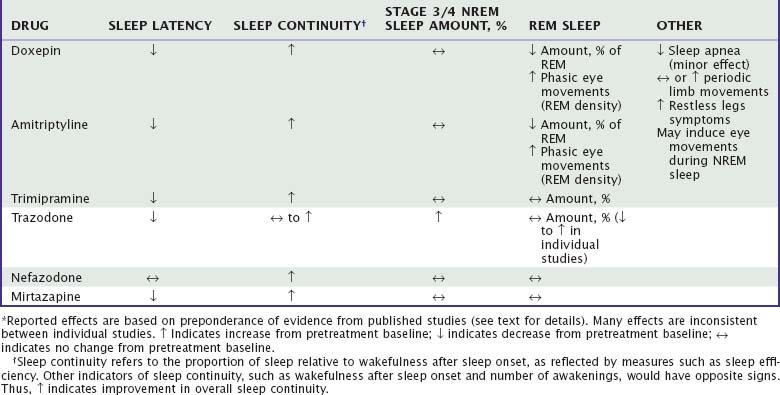
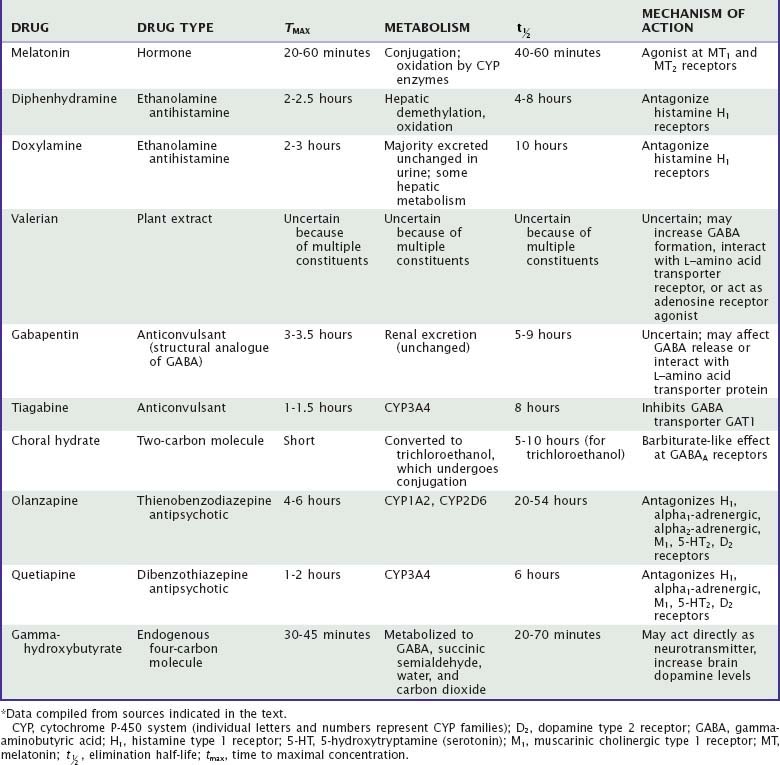
Unlike benzodiazepine receptor agonists, other drugs used to treat insomnia do not share consistent chemical structures, receptor activities, or other pharmacologic properties. Moreover, most nonapproved hypnotic drugs have not been rigorously evaluated with regard to their sleep effects, leaving uncertainties about appropriate dose, demonstrated efficacy, and side effects. Sedating antidepressants and their effects on sleep are summarized in Tables 43-1 to 43-3. The effects of other drugs used as hypnotics are summarized in Tables 43-4 and 43-5.
A Model of Sleep–Wake Regulation Relevant to Sleep-Promoting Drugs
Recent findings from the basic and clinical neuroscience of sleep–wake regulation permit specific models of sleep–wake regulation and insomnia to be advanced and tested (see also Chapter 78). The model in Figure 43-1 describes non–rapid eye movement (NREM) sleep and wakefulness in humans as the products of dynamic interactions among several neural systems.5–10 Wakefulness and arousal states are generated by the ascending activity of monoaminergic brainstem nuclei, histaminergic nuclei in the posterior hypothalamus, and cholinergic nuclei of the pontine tegmentum and basal forebrain (a to c). Activity of these arousal systems is promoted by input from orexin (hypocretin) neurons in the perifornical lateral hypothalamus (LHA) during wakefulness and inhibited by gamma-aminobutyric acid (GABA) and galaninergic neurons in the ventrolateral preoptic area (VLPO) and median preoptic area (MnPO) of the hypothalamus at the onset of sleep (c). The LHA and VLPO/MnPO are in dynamic equilibrium with each other to ensure stable sleep–wake states (d). The timing and overall level of activity in the LHA and VLPO are tightly regulated by homeostatic sleep drive and the circadian timing system (e), which also indirectly affect output of brainstem arousal centers (f). Descending inputs from cortical-diencephalic cognitive and affective centers further modulate sleep–wake balance (g) and are in turn modulated by ascending input from arousal centers, both directly and through thalamic pathways (a, b). Finally, corticothalamic oscillations underlie the characteristic electrophysiologic events of NREM sleep, sleep spindles, and delta oscillations.
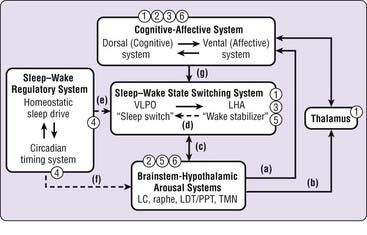
Findings from human functional neuroimaging studies are consistent with this model. Compared with wakefulness, NREM sleep is associated with decreased global blood flow and glucose metabolism as well as with large regional decreases in dorsolateral and medial prefrontal cortex, amygdala and cingulate cortex, thalamus, and brainstem-hypothalamic centers.11–13 The influence of the circadian timing system is suggested by diurnal variation in regional metabolism during wakefulness in humans, with increased brainstem-hypothalamic activity in the morning relative to the evening.14 Finally, increasing homeostatic sleep drive with total sleep deprivation leads to large reductions in cortical, diencephalic, and brainstem structures during wakefulness and sleep.15–17
Sleep-promoting medications may affect sleep–wake regulatory systems at several levels, as indicated in Figure 43-1. Benzodiazepine receptor agonists (1) may directly affect the sleep–wake state-switching system but also have direct cortical, thalamic, and brainstem effects. Sedating antidepressant and antipsychotic medications (2), through their activity on monoaminergic systems, are most likely to affect corticolimbic systems and brainstem-hypothalamic arousal systems. The effects of antihistamines (3) are most likely to result from antagonism of histamine receptors in the hypothalamus and cortex that receive projections from the tuberomammillary nucleus. Melatonin and melatonin receptor agonists (4), through their effects on MT1 and MT2 receptors, influence the “wake signal” from the suprachiasmatic nucleus and circadian timing system. Investigational orexin antagonists (5) are likely to inhibit the effect of orexin-hypocretin on brainstem-hypothalamic arousal centers, and 5-HT2 antagonists (6) are most likely to have corticolimbic and brainstem sites of action. Thus, it seems likely that different types of sleep-promoting drugs achieve their effects through very different actions on very different components of the sleep–wake regulatory system.
Sedating Antidepressants
Overview
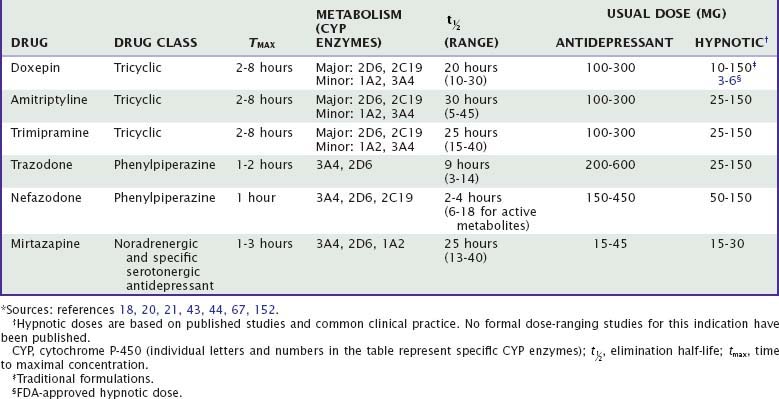
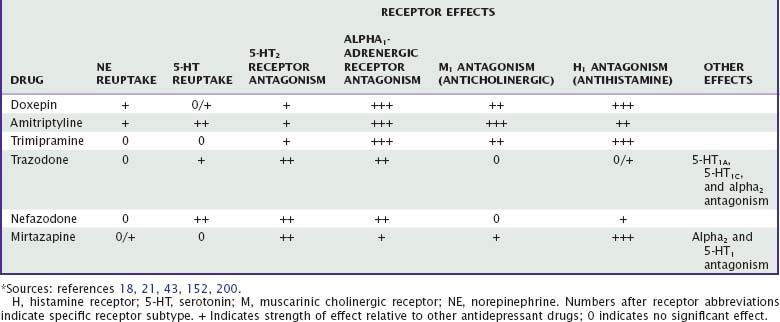
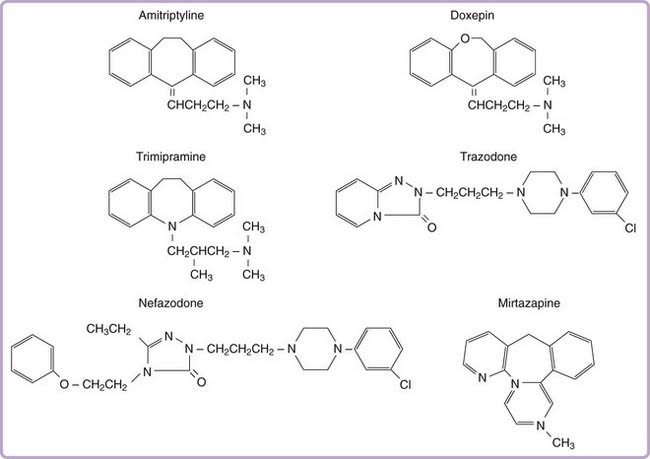
More than two dozen drugs are FDA approved as antidepressants in the United States, but several of these are used, mainly in low doses, for the treatment of insomnia. These include the tricyclic antidepressants (TCAs) doxepin, trimipramine, and amitriptyline as well as the heterocyclic drugs trazodone and mirtazapine. Low-dose doxepin (3 mg and 6 mg) was approved by the FDA for the treatment of insomnia in March, 2010; other sedating antidepressants have not been approved for this indication. The chemical structures of sedating antidepressants are shown in Figure 43-2, and pharmacokinetic properties are summarized in Table 43-1. The complex receptor effects of sedative antidepressants are summarized in Table 43-2.
The majority of data regarding the effects of antidepressant drugs on human sleep come from studies in patients with depression, with use of doses higher than those typically used for insomnia. Formal dose ranging studies have not been conducted to determine optimal hypnotic doses, except for doxepin. Sleep effects of sedating antidepressant drugs have been discussed elsewhere18,19 and are summarized in Table 43-3.
Tricyclic Antidepressant Drugs
Tricyclic antidepressants share a core cyclic structure and differ from one another in their specific side chains (see Fig. 43-2). TCAs are often subgrouped according to side chain structures as tertiary or secondary amines. Tertiary amine TCAs are metabolized to secondary amine TCAs. Both the parent drugs and their metabolites have measurable blood levels and pharmacologic activity. Tertiary TCAs are generally more sedating than secondary TCAs.
Pharmacokinetics
Tricyclic antidepressants are absorbed moderately quickly from the gastrointestinal tract, with maximum concentrations occurring at 2 to 6 hours after the dose. They undergo extensive (30% to 90%) first-pass metabolism and are extensively protein bound (up to 95%). Consequently, bioavailability is low after oral administration.18,20,21 TCAs are lipophilic, ensuring large volumes of distribution and high concentrations in the brain. TCAs have half-lives of approximately 15 to 30 hours.
Tricyclic antidepressants undergo extensive hepatic metabolism, including oxidative demethylation of the side chains and hydroxylation of the ring structure,18–20 followed by conjugation and renal excretion. Hepatic metabolism of TCAs primarily involves CYP2C19 and CYP2D6 as well as CYP1A2 and CYP3A4.19–21 At usual doses, metabolism of TCAs follows linear kinetics, that is, drug blood levels (and metabolism) are proportional to dose; but at higher doses, metabolic enzymes can become saturated, leading to nonlinear pharmacokinetics, that is, higher blood levels than predicted by dose alone.19 Wide interindividual variability in metabolism of TCAs is due to population variability in CYP2D6 polymorphisms and activity.22 Age is associated with decreased metabolic clearance by CYP3A4 and decreased renal clearance.18,20
CYP3A4 can be induced by drugs such as barbiturates and tamoxifen, leading to reduced TCA levels; CYP2D6 can be inhibited by drugs such as antipsychotic drugs, methylphenidate, fluoxetine, and paroxetine, leading to increased TCA levels.19 Grapefruit juice is a common if unsuspected inhibitor of CYP3A4.
Pharmacodynamics and Receptor Pharmacology
Tricyclic antidepressants interact with a variety of neurotransmitter receptors, including serotonin, norepinephrine, acetylcholine, and histamine. Effects on sleep probably represent the combined effects of many of these actions. Amitriptyline and doxepin inhibit both serotonin and norepinephrine reuptake transporters, whereas trimipramine has minimal reuptake effects. Tertiary TCAs including amitriptyline and doxepin have relatively more pronounced effects on serotonin than on norepinephrine reuptake, whereas their secondary amine metabolites have more pronounced effects on norepinephrine reuptake.19,23 After chronic dosing, additional effects on serotonergic and noradrenergic neurotransmission are also observed: desensitization of presynaptic 5-HT1A autoreceptors; upregulation (sensitization) of postsynaptic 5-HT1A receptors; and both downregulation and antagonism of postsynaptic 5-HT2 receptors.24 At 5-HT2 receptors, antagonism of the 5-HT2C subtype is more strongly associated with increasing slow-wave sleep compared with 5-HT2A receptors.25 The net effect of TCA receptor effects is an enhancement of 5-HT effects in the central nervous system (CNS). Noradrenergic effects of TCAs include desensitization of presynaptic alpha2 autoreceptors and a compensatory downregulation of postsynaptic beta receptors,18 with a net effect of increasing noradrenergic neurotransmission. TCAs also antagonize peripheral alpha1– and alpha2-adrenergic receptors, which accounts for their cardiovascular effects.
Effects on Human Sleep
Subjectively, tertiary tricyclic drugs are perceived as sedating, associated with reports of decreased sleep latency and wakefulness during the sleep period and improved sleep quality among depressed patients. Secondary TCAs such as desipramine are less sedating and may even be subjectively alerting. The polysomnographic (PSG) effects of sedating TCAs in depression have been studied extensively. Reduced sleep latency, reduced wakefulness during sleep, and increased sleep efficiency have been reported in depressed patients treated with amitriptyline,26–28 doxepin,29 and trimipramine30,31 and in primary insomnia patients treated with doxepin32,33 and trimipramine.34 By contrast, secondary TCAs such as desipramine and nortriptyline have little or no effect on sleep onset and continuity measures in depressed patients.27,35
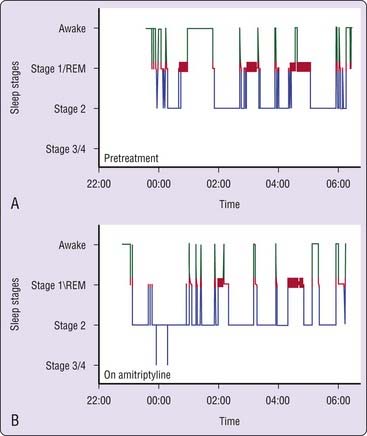
Doxepin and amitriptyline have consistent effects on sleep stage architecture, including reductions in rapid eye movement (REM) sleep percentage and increases in phasic REM activity and REM sleep latency.26,28,29,36,37 Trimipramine differs from most TCAs in its effects on REM sleep, which has been reported to increase, to decrease, or to remain unchanged during treatment.28,30,31,34 Doxepin, amitriptyline, and trimipramine have inconsistent effects on stage 3/4 NREM, which has been reported to increase in some studies37 but not in most others.28,30,31,33 The effects of a TCA on PSG sleep are illustrated in Figure 43-3.
In early studies of insomnia patients, relatively low doses of doxepin (25 to 50 mg) and trimipramine (50 to 200 mg) were associated with improved overall subjective sleep quality and daytime well-being compared with placebo.33,34 PSG effects of doxepin include reduced wakefulness and sleep latency and increased sleep time and sleep efficiency.38 A short-term study of doxepin (1, 3, and 6 mg) in primary insomnia demonstrated reduced wakefulness after sleep onset, wake time during sleep, and (at the highest dose) sleep latency as well as increased sleep time and sleep efficiency.39,39a Sleep efficiency was increased in each third of the night and each hour of the night, but with no demonstrable effect on next-day alertness or psychomotor performance. Doxepin 3 mg and 6 mg was approved by the FDA in March, 2010 for the treatment of insomnia characterized by sleep maintenance difficulty. Trimipramine has been reported to reduce wakefulness and improved sleep efficiency without affecting sleep time or sleep latency.40
Other effects of TCAs on PSG sleep can include increases in periodic limb movements during sleep and the appearance of eye movements during NREM sleep; such effects are particularly noted with strongly serotonergic TCAs such as clomipramine. TCAs do not worsen sleep apnea and may have a small beneficial effect.36 The acute effects of TCAs on sleep in depression are maintained during 1 to 3 years of maintenance treatment.36,41 Rebound insomnia, indicated by decreased sleep time and sleep efficiency, may occur on discontinuation of sedating TCAs.42
Side Effects
Anticholinergic side effects of doxepin and amitriptyline include dry mouth, increased perspiration, constipation, and urinary retention. More serious effects include precipitation of ocular crises in patients with narrow-angle glaucoma, seizures, and anticholinergic delirium, which are dose related and typically occur at blood levels above 300 ng/mL.18 Side effects related to antihistaminic properties include sedation and weight gain. Studies of very low dose doxepin (1 to 6 mg) show a low rate of side effects, with somnolence being the most common.39
Side effects related to alpha1 antagonism include orthostatic hypotension with attendant risks of lightheadedness, syncope, and falls. TCAs typically increase heart rate. They also slow cardiac electrical conduction due to type I antiarrhythmic (quinidine-like) effects, which can lead to prolongation of the QRS duration and PR and QT intervals and heart block. TCA overdose lethality is largely due to cardiovascular toxicity, which can occur at doses as low as 10 times the therapeutic antidepressant daily dose. On the other hand, TCAs have no effect on cardiac contractility, and they can suppress atrial and ventricular ectopy.18
Trazodone and Nefazodone
Pharmacokinetics
Trazodone is rapidly absorbed, with peak plasma concentrations occurring 1 to 2 hours after oral doses. Like TCAs, it is highly (85% to 95%) protein bound. Nefazodone is rapidly and completely absorbed but rapidly metabolized, resulting in low bioavailability (about 20%). Trazodone and nefazodone have short half-lives of approximately 5 to 9 hours and 2 to 4 hours, respectively. However, active metabolites of nefazodone have slightly longer half-lives.43,44
The major metabolic pathway for trazodone and nefazodone is N-dealkylation to produce m-chlorophenylpiperazine (mCPP), an active metabolite that possesses serotonergic activity.21,43 Both drugs also undergo oxidation. Trazodone and mCPP are substrates for CYP2D6, and trazodone is also metabolized to a lesser extent by CYP3A4. Nefazodone is oxidized by CYP3A4 and CYP2C19. Drugs that inhibit CYP3A4, such as ketoconazole, inhibit trazodone and nefazodone metabolism and decrease mCPP formation.43 Nefazodone and hydroxynefazodone themselves can also inhibit the activity of CYP3A4, thereby impairing the clearance and raising the plasma level of other drugs metabolized by the same route, including alprazolam, diazepam, and triazolam.45 Nefazodone metabolism and hepatic clearance are also reduced by age and liver impairment.
Pharmacodynamics and Receptor Pharmacology
Trazodone is a relatively weak but specific inhibitor of the serotonin reuptake transporter with minimal affinity for norepinephrine or dopamine reuptake. Trazodone also inhibits serotonin 5-HT1A, 5-HT1C, and 5-HT2 receptors. It has essentially no affinity for M1 receptors, but it does have moderate H1 histamine receptor antagonism. Finally, trazodone is a relatively weak antagonist of alpha2-adrenergic receptors and a somewhat more potent antagonist of alpha1 receptors.21,43
Like trazodone, nefazodone is a serotonin 5-HT2 receptor antagonist and a weak inhibitor of both serotonin and norepinephrine reuptake. Unlike trazodone, nefazodone shows little affinity for serotonin 5-HT1A receptors; adrenergic alpha1, alpha2, or beta receptors; or histamine H1 receptors.43
The active metabolite mCPP inhibits serotonin reuptake and is a partial agonist at postsynaptic 5-HT2C receptors. These actions can lead to increased side effects from trazodone and nefazodone among individuals who are “poor metabolizers” through CYP2D6 or who are taking inhibitors of CYP2D6, such as fluoxetine.44 Nefazodone shows nonlinear pharmacokinetics, resulting in greater than proportional plasma levels after higher doses.43
Effects on Human Sleep
Studies of trazodone effects on human sleep have been limited by small sample sizes, particularly in PSG studies, and by study designs that have typically included sleep only as a secondary endpoint. In addition, many studies specifically addressing the sleep effects of trazodone have not included double-blind, placebo-controlled, randomized study designs. No published study has examined PSG outcomes with trazodone in patients with primary insomnia.46 Sedation is a common effect of trazodone in the treatment of depression, reported by more than 40% of patients. When it is specifically assessed, subjective sleep quality is improved by trazodone in healthy controls47 and in patients with depression48–51 and alcohol dependence.52 A study of sustained-release trazodone versus sertraline showed greater improvement in sleep disturbances with trazodone.53 However, some negative studies with respect to sleep quality in depressed subjects have also been reported.54
Polysomnographic studies have not consistently reported the effects of trazodone on sleep continuity. The available evidence is inconsistent with regard to effects on sleep latency, total sleep time, and sleep efficiency; about half of the published studies show improvements in these measures.53 Trazodone improved sleep efficiency, total sleep time, and wakefulness during sleep in older controls,47 in patients with depression49,55,56 (including depressed patients concurrently treated with selective serotonin reuptake inhibitors51), and in abstinent alcohol-dependent patients.57 Some negative studies with regard to sleep continuity have also been reported in younger healthy controls,58 in which ceiling effects may have been an issue, and in some studies of depressed patients with small numbers of subjects.48,54
Unlike most TCAs, trazodone has little effect on the amount of REM sleep; a majority of studies show no significant change49,55,56,58 or a small decrease.47,54 Trazodone is also associated with increased stage 3/4 NREM sleep in a majority of studies.47–49,51,56,58 In one study, a small reduction in apnea-hypopnea index and no change in periodic limb movements were noted.49 Although no information has been published about its long-term effects on sleep, rebound insomnia has been noted on discontinuation after several weeks of use.47
Only one study has been published on the effects of trazodone in primary insomnia compared with both placebo and zolpidem.59 Compared with placebo, trazodone (50 mg) was associated with reduced subjective sleep latency, awakenings, and wake time during sleep and increased subjective sleep time, ease of falling asleep, and sleep quality. These findings appeared to persist for both study weeks, but significant differences versus placebo were noted only during the first week because of late improvements in the placebo condition. The magnitude of effects with trazodone was similar to that for zolpidem (10 mg).
Nefazodone is less consistently sedating than trazodone. A series of studies in depressed patients comparing nefazodone and fluoxetine showed that both drugs improved subjective sleep disturbance, but nefazodone had a significantly greater effect.60–62 Nefazodone was associated with increased PSG sleep efficiency, decreased wakefulness during sleep, and no change in sleep latency. REM sleep was increased in two of the studies and unchanged in one, and stage 3/4 was decreased in two studies and increased in one. Thus, nefazodone appears to have somewhat less potent effects on either sleep continuity or slow-wave sleep than trazodone has. No published studies have specifically examined nefazodone in patients with primary insomnia.
Side Effects
Trazodone can produce side effects including orthostatic hypotension, lightheadedness, and weakness.43 Unlike TCAs, trazodone does not have anticholinergic side effects, but it can have antihistaminic effects such as weight gain. Case reports suggest a potential for ventricular tachyarrhythmias.63 A potentially serious effect of trazodone is priapism, sustained painful erections in men.64 Although it is uncommon, priapism can be serious, requiring prompt surgical treatment. The risk appears to be greatest early in the course of treatment, and it can occur even at low doses. The incidence of abnormal erections during trazodone treatment is approximately 1 per 6000 male patients treated. Fatalities have been reported with overdoses of trazodone, although most of these occurred in conjunction with other drug ingestion. Unlike zolpidem and triazolam, trazodone is not associated with subjective effects associated with abuse potential.65
Common side effects associated with nefazodone include dry mouth, somnolence, dizziness, nausea, blurred vision, and postural hypotension.43 Nefazodone has been associated with serious hepatic toxicity, in some cases leading to fulminant hepatic failure and death.66 The reported rate of life-threatening liver failure is estimated at 1 case per 250,000 to 300,000 patient-years of exposure. This risk has led to withdrawal of nefazodone from the market in some countries. On the other hand, nefazodone has not been associated with fatal toxicity in overdose. There has been concern that mCPP, the metabolite of trazodone and nefazodone, can contribute to the development of the “serotonin syndrome” when this drug is used in combination with other serotonergic drugs, such as selective serotonin reuptake inhibitors. The serotonin syndrome includes symptoms of confusion or delirium, restlessness similar to akathisia, muscle irritability, hyperreflexia, and autonomic instability including hypotension.
Mirtazapine
Pharmacokinetics
Mirtazapine is rapidly absorbed, undergoes extensive first-pass metabolism, and is about 85% protein bound, yielding bioavailability of about 50%.67,68 Mirtazapine has an elimination half-life of approximately 20 to 40 hours.
Mirtazapine undergoes N-demethylation (producing an active metabolite) and N-oxidation, followed by conjugation and excretion. CYP2D6 and, to a lesser extent, CYP3A4 and CYP1A2 are the major enzymes involved in mirtazapine metabolism. Mirtazapine follows linear pharmacokinetics, but its metabolism is affected by both age and sex; metabolic clearance is reduced in women and in older adults as well as in those with liver disease.67 Although mirtazapine itself does not strongly inhibit or induce hepatic enzymes, mirtazapine blood levels are decreased by medications such as carbamazepine that induce CYP enzymes and increased by medications such as fluoxetine that inhibit CYP enzymes.
Pharmacodynamics and Receptor Pharmacology
Mirtazapine is a very weak inhibitor of noradrenergic reuptake and has no effect on serotonin reuptake.69 However, similar to TCAs, it increases serotonergic and noradrenergic neurotransmission through blockade of alpha2 autoreceptors and heteroreceptors.44 It also has prominent antagonist activity at 5-HT2, 5-HT3, H1, and alpha1-adrenergic receptors,69 which may contribute to its hypnotic effects.
Effects on Human Sleep
Mirtazapine is reported to be subjectively sedating in clinical studies of depression. In a PSG study in healthy adults, mirtazapine decreased sleep latency, awakenings, and stage 1 NREM sleep and increased stage 3/4.70 In a study of six depressed patients, mirtazapine had similar effects, decreasing sleep latency, increasing sleep efficiency and sleep time, and having no significant effect on REM or stage 3/4 sleep.71 Thus, although it is potentially promising, mirtazapine has not yet been adequately evaluated as a hypnotic.
Side Effects
In addition to causing sedation, mirtazapine is associated with increased appetite, weight gain, and dry mouth, probably related to its antihistaminic properties. Clinical observations suggest that mirtazapine may be less sedating at doses above 30 mg per day than at lower doses. This is hypothesized to be related to greater noradrenergic effects relative to antihistaminic and serotonergic effects at lower doses.68 Mirtazapine has not been associated with serious toxicity or death in overdose.
Serotonin Receptor (5-Ht2a/2c) Antagonists
As discussed before, several antidepressants are antagonists of serotonin 5-HT2 receptors, which is thought to underlie some of their sedative effect. Other compounds that antagonize 5-HT2A and 5-HT2C receptors have been investigated more specifically for their sleep effects, although some of them have also been investigated as antidepressant or antianxiety drugs as well. The role of serotonin in sleep–wake regulation was a central tenet of the monoamine theory of sleep and wakefulness,72 stemming from the observation that destruction of serotonin neurons in the dorsal raphe, or depletion with reserpine, results in severe insomnia. Subsequent observations showed serotonergic raphe neurons to be most active during wakefulness, less active during NREM sleep, and virtually inactive during REM sleep.73 The large number of serotonin actions and receptor subtypes makes it difficult to propose any general function of this widespread neurotransmitter. 5-HT2A/2C receptors are G protein–coupled receptors that are widely distributed in the CNS, including the cortex, hippocampus, amygdala, thalamus, hypothalamus, and brainstem. They functionally interact with and affect the activity of GABAergic, dopaminergic, and cholinergic neurons.74
Early evidence in healthy humans showed that ritanserin, a 5-HT2A/2C antagonist, increased slow-wave sleep and delta electroencephalographic (EEG) activity in a dose-dependent fashion, with inconsistent and smaller magnitude effects on subjective sleep quality and PSG sleep latency and awakenings.75–78 A comparison with ketanserin, a more selective 5-HT2A antagonist, suggested that the effects of ritanserin on 5-HT2C receptors were more likely to explain its slow-wave sleep-enhancing actions.78 Another 5-HT2 antagonist, SR 46349B, was observed to have effects similar to those of ritanserin, increasing visually scored slow-wave sleep and suppressing REM sleep.79 This agent also increased slow-wave activity and reduced spindle frequency activity on quantitative EEG measures. Ritanserin was found to have few effects on sleepiness or psychomotor function, even with daytime administration. Among poor sleepers, ritanserin had a small effect on the number of awakenings, in addition to the previously described slow-wave sleep effect and improved subjective sleep quality.80,81 Acute discontinuation led to rebound insomnia.
Although ritanserin was not further developed as a hypnotic medication, several new 5-HT2 antagonists are undergoing clinical trials for this indication. The first published study with APD-125 demonstrated reduced wakefulness after sleep onset and wakefulness during sleep, increased slow-wave sleep, and improved subjective sleep, but no effect on sleep latency, in patients with chronic insomnia.82
Melatonin and Melatonin Receptor Agonists
Biosynthesis, Physiologic Regulation, and Specific Compounds
As reviewed elsewhere in this volume, melatonin is a hormone endogenously synthesized from serotonin and produced in the pineal gland, retina, and intestinal tract. Melatonin production is acutely suppressed by light acting on the retina. The noradrenergic sympathetic nervous system, acting through the superior sympathetic ganglion, stimulates pineal melatonin production. Specifically, beta1 stimulation with alpha1 amplification leads to increased availability of N-acetyltransferase, the rate-limiting enzyme in melatonin biosynthesis.83 Hence, beta- and alpha-adrenergic antagonists may affect melatonin synthesis. A number of synthetic melatonin receptor agonists have been developed and tested in laboratory and clinical studies. These include ramelteon and agomelatine (S-20098), a serotonergic-melatonergic antidepressant.84,85

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


