Epilepsy and Other Seizure Disorders: Introduction
The prevalence and importance of epilepsy, i.e., recurrent cerebral cortical seizures, can hardly be overstated. From the epidemiologic studies of Hauser and colleagues, one may extrapolate an incidence of approximately 2 million individuals in the United States who are subject to epilepsy and predict about 44 new cases per 100,000 persons each year. These figures are exclusive of patients in whom seizures transiently complicate febrile and other illnesses or injuries. It has also been estimated that slightly less than 1 percent of persons in the United States will have epilepsy by the age of 20 years (Hauser and Annegers). Over two-thirds of all epileptic seizures begin in childhood (most in the first year of life), and this is the period when seizures assume the widest array of forms. In the practice of pediatric neurology, epilepsy is one of the most common disorders, and the chronicity of childhood forms adds to their importance. The incidence increases again after age 60 years. For all these reasons, physicians should know something of the nature of seizure disorders and their treatment. It is notable that, in striking contrast to the many treatments available for epilepsy, as pointed out by J. Engel, 80 to 90 percent of persons with epilepsy in the developing world never receive medical attention.
In 1870, Hughlings Jackson, the eminent British neurologist, postulated that seizures were due to “an excessive and disorderly discharge of cerebral nervous tissue on muscles.” The discharge may result in an almost instantaneous loss of consciousness, alteration of perception or impairment of psychic function, convulsive movements, disturbance of sensation, or some combination thereof.
Terminologic difficulty arises from the diversity of the clinical manifestations of seizures. The term convulsion, referring as it does to an intense paroxysm of involuntary repetitive muscular contractions, is inappropriate for a disorder that may consist only of an alteration of sensation or consciousness. Seizure is preferable as a generic term, because it embraces all paroxysmal electrical discharges of the brain, and also because it lends itself to qualification. The term motor or convulsive seizure, therefore, is not tautologic, and one may likewise speak of a sensory seizure or psychic seizure. The word epilepsy is derived from Greek words meaning “to seize upon” or a “taking hold of.” Our predecessors referred to it as the “falling sickness” or the “falling evil.” Although a useful medical term to denote recurrent seizures, the words epilepsy and epileptic may still have unpleasant connotations and should be used advisedly in dealing with patients. There is also a curious, but common entity of “nonconvulsive seizure” that may impair consciousness, but not manifest any abnormal bodily movement. This represents an important and potentially treatable form of a confusional state.
A first solitary seizure or brief outburst of seizures may occur during the course of many medical illnesses. It indicates that the cerebral cortex has been affected by disease, either primarily or secondarily. If prolonged or repeated every few minutes, the condition termed status epilepticus, may threaten life. Equally important, a seizure or a series of seizures may be the manifestation of an ongoing neurologic disease that requires special diagnostic, and therapeutic measures. Status epilepticus may be of the nonconvulsive type, and continuously impair consciousness and is difficult to detect clinically because of the absence of characteristic movements.
A more common and less-grave circumstance is for a seizure to be but one in an extensive series recurring over a long period of time, with most of the attacks being more or less similar in type. In this instance, they may be the result of an inactive lesion that remains as a scar in the cerebral cortex. The original disease may have passed unnoticed, or perhaps had occurred in utero, at birth, in infancy, or in parts of the brain inaccessible for examination or too immature to manifest signs. The increasingly refined techniques of MRI now also reveal small zones of developmental cortical dysplasia and hippocampal sclerosis, both of which tend to be epileptogenic. Patients with such long-standing but subtle lesions probably make up a large portion of those with recurrent seizures. If there is no underlying lesion, the condition is classified as idiopathic or primary, but in the modern era, this is come to be almost synonymous with a genetic cause. In this category, there are a large number of important types of epilepsy for which no pathologic basis has been established, and for which there is no apparent underlying cause except perhaps a genetic one. Included here are special hereditary forms including types of generalized tonic-clonic (grand mal), and “absence” seizure states as suggested in classifications many years ago by Lennox and Forster. Persistent seizures, whether idiopathic or not, can secondarily damage cortical tissue by several mechanisms that may include excitotoxicity and, in the setting of prolonged tonic seizures, systemic hypoxia.
Classification of Seizures and Epilepsies
Seizures have been grouped in several ways: according to their presumed etiology, i.e., idiopathic (primary) or symptomatic (secondary); their site of origin; their clinical form (generalized or focal); their frequency (isolated, cyclic, or repetitive, or the closely spaced sequence of status epilepticus); or by special electrophysiologic correlates. A distinction must be made between the classification of seizures (the clinical manifestations of epilepsy: generalized tonic clonic (grand mal), absence (petit mal), myoclonic, partial, and others), and the classification of the epilepsies, or epileptic syndromes, which are specific diseases, most of which may manifest several seizure types. These are discussed later in the chapter. A further distinction is made by clinical and EEG features. This approach allows for the reasonable predictability of response to specific medications and to some extent, in prognosis. Basically, this classification divides seizures into two types—focal (formerly termed partial), in which a focal or localized onset can be discerned clinically or by EEG, and generalized, in which the seizures appear to begin bilaterally.
Generalized seizures are of two types—convulsive and nonconvulsive. (This dichotomy is not part of the main classification, but it is fundamental.) The common convulsive type is the tonic-clonic (grand mal) seizure. Less common is a purely tonic, purely clonic, or clonic-tonic-clonic generalized seizure. The typical nonconvulsive generalized seizure is the brief lapse of consciousness or absence (petit mal); included also under this heading are minor motor phenomena such as brief myoclonic, atonic, or tonic seizures.
The classification followed here was first proposed by Gastaut in 1970 and has been refined repeatedly by the Commission on Classification and Terminology of the International League Against Epilepsy. This nomenclature, based mainly on the clinical form of the seizure, and its electroencephalographic (EEG) features, has been adopted worldwide and is generally referred to as the “International Classification”. A modified version of it is reproduced in Table 16-1.
I. Generalized seizures (bilaterally symmetrical and without focal onset) |
A. Tonic, clonic, or tonic-clonic (grand mal) |
B. Absence (petit mal) |
1. typical |
2. atypical |
3. special features |
a. eyelid myoclonia |
b. myoclonic absence |
C. Clonic |
D. Tonic |
E. Atonic |
F. Myoclonic including atonic and tonic types |
II. Focal (formerly “partial”); characterized by main feature(s). See Table 16-2 |
A. Simple (without loss of consciousness or alteration in -psychic function) |
1. Aura; somatosensory or special sensory (visual, auditory, olfactory, gustatory, vertiginous) |
2. Motor |
3. Autonomic |
4. Awareness retained (formerly “simple”) or impaired (formerly “complex”) |
III. Unclassifiable; cannot be characterized as focal, generalized or both, including epileptic spasms |
CLINICAL TYPE | LOCALIZATION |
|---|---|
Somatic motor | |
Jacksonian (focal motor) | Prerolandic gyrus |
Masticatory, salivation, speech arrest | Amygdaloid nuclei, opercular |
Simple contraversive | Frontal |
Head and eye turning associated with arm movement or athetoid-dystonic postures | Supplementary motor cortex |
Somatic and special sensory (auras) | |
Somatosensory | Contralateral postrolandic |
Unformed images, lights, patterns | Occipital |
Auditory | Heschl gyri |
Vertiginous | Superior temporal |
Olfactory | Mesial temporal |
Gustatory | Insula |
Visceral: autonomic | Insular-orbital-frontal cortex |
Focal seizure with altered consciousness | |
Formed hallucinations | Temporal neocortex or amygdaloid–hippocampal complex |
Illusions | — |
Dyscognitive experiences (déjà vu, dreamy states, depersonalization) | — |
Affective states (fear, depression, or elation) | Temporal |
Automatism (ictal and postictal) | Temporal and frontal |
Staring | Frontal cortex, amygdaloid–hippocampal complex, reticular–cortical system |
It is also useful clinically, and etiologically to separate epilepsies that originate as truly generalized electrical discharges in the brain from those that spread secondarily from a focus to become generalized. The primary generalized epilepsies are a group of somewhat diverse, age-dependent phenotypes that are characterized by generalized 2.5- to 4-Hz bifrontally predominant spikes or polyspike-and-slow-wave discharges that arise without underlying structural abnormalities. In most instances, these individuals have normal intelligence. What is most significant is that a genetic component underlies many of these disorders. By contrast, epilepsies manifesting as seizures that begin locally and may evolve into generalized tonic-clonic seizures, termed secondarily generalized tonic clonic seizures (termed bilateral convulsive in Fig. 16-1), generally have no such genetic component and are usually the result of underlying brain disease, either acquired or a result of congenital malformations or metabolic defects. Quite often, the initial focal phase is not appreciated, leading to misdiagnosis. An increasing frequency and severity of this group of disorders with age reflects the accumulation of focal cerebral damage from trauma, strokes, and other damage.
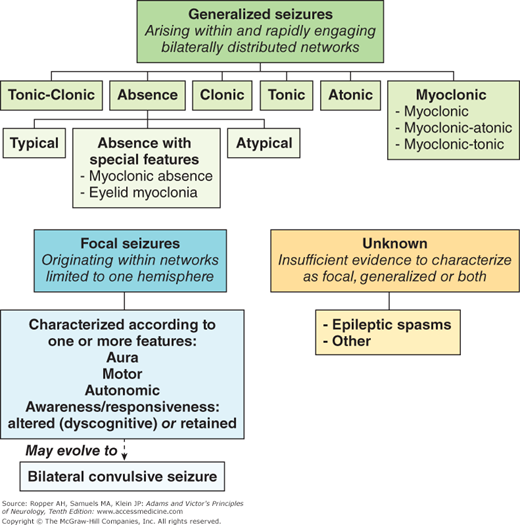
Focal seizures are further classified according to their additional features such as a specific subjective experience (aura), motor, autonomic, and most importantly, whether awareness or consciousness is disturbed; the latter was formerly called partial complex seizure. A prominent component of such seizures is a dyscognitive state. In reality, an aura represents the initial phase of a focal seizure; in some instances, it may constitute the entire epileptic attack.
The classification of seizures and of the epilepsies is constantly being modified. In an older but still useful version, the so-called syndromic classification (Epilepsia 30:389, 1989), an attempt had been made to incorporate all of the seizure types and epileptic syndromes and to categorize them not only as partial and generalized but also according to their age of onset, their primary (generalized) or secondary nature, the evidence of cortical loci of the epileptogenic lesions, and the many clinical settings in which they occur. This classification is semantically difficult and, in our view, too complicated for general clinical application; it has been replaced with current classifications already mentioned. Because many epileptic syndromes share overlapping features, it is often not possible to fit a newly diagnosed case of epilepsy into a specific category in this new classification. The Commission is continuously engaged in revision of terminology and classification in the field of epilepsy.
We begin our discussion with a practical approximation of the most recent classification that was given in 2010 (see Berg and colleagues) and is shown in Fig. 16-1, followed by consideration of a number of well-defined epilepsies and epileptic syndromes.
It is also useful to view the various types of seizures and epilepsies in the context of the age at which they occur. A proposed current classification based on the age of onset of the seizure disorder is shown in Fig. 16-2, and the distribution of the seizure types for each age epoch, obtained and aggregated from several sources is shown in Fig. 16-3. There has also been substantial progress in defining the molecular basis of familial and hereditary epilepsies over the last decade; it is likely that these insights will lead to modification of both the clinical classifications and management of the epilepsies.
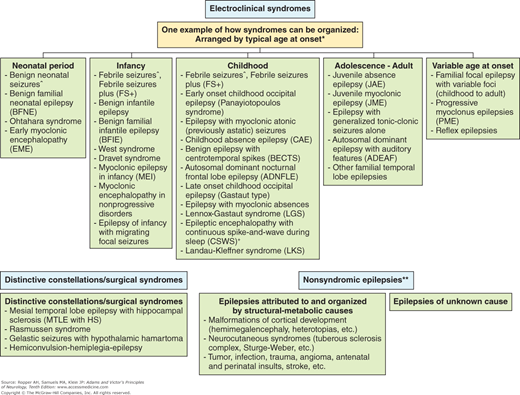
Figure 16-3.
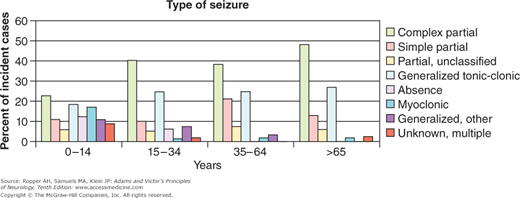
The distribution of the main types of epilepsy by age. The overrepresentation of absence and myoclonic seizures in childhood and of complex partial seizures in older individuals is evident. (Adapted from Hauser and Annegers and the texts of Engel and of Pedley.)
Generalized Seizures
In the common primary type of seizure, most often a convulsion starts with little or no warning. Sometimes the patient senses the approach of a seizure by several subjective phenomena (prodrome) even prior to an epileptic aura, which represents a focal seizure. For some hours, the patient may feel apathetic, depressed, irritable, or, rarely, the opposite—ecstatic. In a patient with generalized epilepsy (juvenile myoclonic epilepsy being one typical type), one or more myoclonic jerks of the trunk or limbs on awakening may herald a seizure later in the day. Abdominal pains or cramps, a sinking, rising, or gripping feeling in the epigastrium, pallor or redness of the face, throbbing headache, constipation, or diarrhea have been given prodromal status, but they do not occur consistently enough to be predictive of an oncoming seizure.
In more than half of cases of generalized seizure, there is some type of movement for a few seconds before consciousness is lost (turning of the head and eyes or whole body or intermittent jerking of a limb), although the patient often fails to form a memory of this and such information is obtained only from an observer. As has already been pointed out, it is useful, whenever possible, to distinguish between a primary (generalized) type of seizure with widespread EEG abnormalities at the onset, and a secondarily generalized type, which begins as a focal seizure and then becomes generalized. The secondary generalized type implicates a focal brain lesion. More often, the seizure strikes without warning, beginning with a sudden loss of consciousness and a fall to the ground that may lead to facial and dental injuries.
The initial motor signs are a brief flexion of the trunk, an opening of the mouth and eyelids, and upward deviation of the eyes. The arms are elevated and abducted, the elbows semiflexed, and the hands pronated. These are followed by a more protracted extension (tonic) phase, involving first the back and neck, then the arms and legs. There may be a piercing cry as the whole musculature is seized in a spasm with biting of the lateral margin of the tongue, and air is forcibly emitted through the closed vocal cords. Because the respiratory muscles are caught up in the tonic spasm, breathing is suspended and after some seconds the skin and lips may become cyanotic. The pupils are dilated and unreactive to light. The bladder may empty at this stage or later, during the postictal coma. This is the tonic phase of the seizure and lasts for 10 to 20 s.
There then occurs a transition from the tonic to the clonic phase of the convulsion. At first, there is a mild generalized tremor, which is, in effect, a repetitive relaxation of the tonic contraction. It begins at a rate of 8 per second and coarsens to 4 per second; then it rapidly gives way to brief, violent flexor spasms that come in rhythmic salvos and agitate the entire body. The face becomes violaceous and contorted by a series of grimaces. Autonomic signs are prominent: the pulse is rapid, blood pressure is elevated, pupils are dilated, and salivation and sweating are prominent; bladder pressure may increase sixfold during this phase. The clonic jerks decrease in amplitude and frequency over a period of about 30 s. The patient remains apneic until the end of the clonic phase, which is often marked by a deep inspiration. Instead of the whole dramatic sequence described above, the seizures may be abbreviated or limited in scope by anticonvulsive medications.
In the terminal phase of the seizure, all movements have ended and the patient is motionless and limp in a deep coma. The pupils now begin to contract to light. Breathing may be quiet or stertorous. This state persists for several minutes, after which the patient opens his eyes, begins to look about, and is obviously bewildered and confused and may be quite agitated. The patient may speak and later not remember anything that has been said and undisturbed becomes drowsy and falls asleep, sometimes for several hours, then often awakens with a pulsatile headache. When fully recovered, such a patient has no memory of any part of the spell but knows that something has happened because of the strange surroundings (in ambulance or hospital), the obvious concern of those around him, and often, a sore, bitten tongue and aching muscles from the violent movements. The contractions, if violent enough, may crush a vertebral body or result in a serious injury; a fracture, periorbital hemorrhages, subdural hematoma, posterior shoulder dislocation, or burn may have been sustained in the fall.
Each of these phases of the generalized tonic-clonic seizure has its characteristic EEG accompaniment. Initially, movement artifacts obscure the EEG tracing; sometimes there are repetitive spikes or spike-wave discharges lasting a few seconds, followed by an approximately 10-s period of 10-Hz spikes. As the clonic phase asserts itself, the spikes become mixed with slow waves and then the EEG slowly assumes a polyspike-and-wave pattern. When all movements have ceased, the EEG tracing is nearly flat for a variable time, and then the brain waves gradually resume their preseizure pattern.
Convulsions of this type ordinarily come singly or in groups of two or three and may occur when the patient is awake and active or during sleep, or when falling asleep or awakening. It is useful to know that seizures on awakening usually signify a generalized type, whereas those occurring during the period of sleep are more often focal in nature. Some 5 to 8 percent of such patients will at some time have a prolonged series of such seizures without resumption of consciousness between them; this is called status epilepticus and demands urgent treatment. The first outburst of seizures may take the form of status epilepticus.
Aside from psychogenic episodes that imitate seizures, few clinical states simulate a generalized tonic-clonic seizure, but several are worthy of mention. One is a clonic jerking of the extended limbs (usually less severe than those of a grand mal seizure) that occurs with vasodepressor syncope or a Stokes-Adams hypotensive attack. In contrast to an epileptic type of EEG, the brain waves are slow in frequency and low in amplitude during the jerking movements. A rarer phenomenon that may be indistinguishable from a generalized convulsion occurs as part of the syndrome of basilar artery occlusion. This presumably has its basis in ischemia of the corticospinal tracts in the pons (Ropper); a similar ischemic mechanism in the cortex has been invoked for “limb-shaking TIAs” (transient ischemic attacks), in which there are clonic movements of one limb or one side of the body during an episode of cerebral ischemia. Clonic limb movements occur immediately after a traumatic concussion and an observer who arrives at this moment will be unable to determine if the inciting event was a seizure or a collision. In infants, a breath-holding spell may closely simulate the tonic phase of a generalized seizure. Another disorder that simulates a seizure, albeit self-induced, is the “fainting lark” (or in the British, the ‘mess trick).” By hyperventilating in a squatting position and standing rapidly combined with a Valsalva maneuver, a syncopal episode is induced that ends with generalized convulsive movements (see Lempert and colleagues).
In contrast to major generalized seizures, absence seizures (formerly referred to as petit mal or pyknoepilepsy) are notable for their brevity, rapid onset and cessation, and frequency and the paucity of motor activity. Indeed, they may be so brief that the patients themselves are sometimes not aware of them; to an onlooker, they resemble a moment of absentmindedness or daydreaming. The attack, coming without warning, consists of a sudden interruption of consciousness, for which the French word absence (“not present,” “not in attendance”) has been retained. The patient stares and briefly stops talking or ceases to respond. Only about 10 percent of such patients are completely motionless during the attack; in the remainder, one observes a brief burst of fine clonic (myoclonic) movements of the eyelids, facial muscles, or fingers or small synchronous movements of both arms, all at a rate of 3 per second. This rate corresponds to that of the EEG abnormality, which takes the form of a generalized 3-per-second spike-and-wave pattern (Fig. 2-3E). Absence seizures are said to be “typical” if they have a rapid onset and offset, typical three per second spike and wave, and complete loss of awareness.
Minor automatisms—in the form of lip-smacking, chewing, and fumbling movements of the fingers—are common during an attack but may be subtle. Postural tone may be slightly decreased or increased, and occasionally there is a mild vasomotor disorder. As a rule, such patients do not fall; they may even continue complex acts such as walking or riding a bicycle. After 2 to 10 s, occasionally longer, the patient reestablishes full contact with the environment and resumes his preseizure activity. Only a loss of the thread of conversation or the place in reading betrays the occurrence of the momentary “blank” period (the absence). In many such patients, voluntary hyperventilation for 2 to 3 min is an effective way of inducing absence attacks.
Typical absence seizures constitute the most characteristic epilepsy of childhood (“childhood absence”); rarely do the seizures begin before 4 years of age or after puberty. Another attribute is their great frequency (hence, the old term pykno, meaning “compact” or “dense”). As many as several hundred may occur in a single day, sometimes in bursts at certain consistent times of the day. They produce periods of inattention and may appear in the classroom when the child is sitting quietly rather than participating actively in his lessons. If frequent, they disturb attention and thinking to the point that the child’s performance in school is impaired. Less frequently, such attacks may last for hours with no interval of normal mental activity between them—so-called absence or petit mal status. Absence epilepsy of adolescent onset (“juvenile absence”) does not have the very high seizure frequency of the childhood type. Cases of absence status have also been described in adults with frontal lobe epilepsy (see below). In contrast to childhood absence seizures, the disorder may last well into adulthood and be punctuated by generalized tonic-clonic seizures or a burst of seizures. Akinesia (motionlessness) is not unique to any seizure type.
The typical absence, with or without myoclonic jerks, rarely causes the patient to fall. Absence should be considered a separate entity because of its relative benignity. It may be the only type of seizure during childhood. The attacks tend to diminish in frequency in adolescence and then often disappear, only to be replaced in many instances by major generalized seizures. About one-third of children with absence attacks will, in addition, display symmetrical or asymmetrical myoclonic jerks without loss of consciousness, and about half will at some time have major generalized (tonic-clonic) convulsions.
Distinguished from typical absence seizures are variants in which the loss of consciousness is less complete or in which myoclonus is prominent, and others in which the EEG abnormalities are less regularly of a 3-per-second spike-and-wave type (they may occur at the rate of 2 to 2.5 per second or take the form of irregular 4- to 6-Hz polyspike-and-wave complexes). Atypical absence is a term that was introduced to describe long runs of slow spike-and-wave activity, usually with no apparent loss of consciousness. External stimuli, such as asking the patient to answer a question or to count, interrupt the run of abnormal EEG activity. The current classification (Fig. 16-1) separates the disorder into groups that are identified as typical, atypical, and having special features, namely, myoclonic jerks or eyelid myoclonus.
In sharp contrast to the typical absence epilepsies, is a form that has its onset between 2 and 6 years of age and is characterized by atonic, or astatic, seizures (i.e., falling attacks), often succeeded by various combinations of minor motor, tonic-clonic, and partial seizures and by progressive intellectual impairment in association with a distinctive, slow (1- to 2-Hz) spike-and-wave EEG pattern. This is the Lennox-Gastaut syndrome. Often it is preceded in earlier life by infantile spasms, a characteristic high-amplitude chaotic EEG picture (“hypsarrhythmia”), and an arrest in mental development, a triad sometimes referred to as the West syndrome (see further on). The early onset of atonic seizures with abrupt falls, injuries, and associated abnormalities nearly always has a grave implication—namely, the presence of serious neurologic disease. Prematurity, perinatal injury and metabolic diseases of infancy are the most common underlying conditions. This is essentially a form of symptomatic generalized epilepsy, in contrast to the foregoing idiopathic types such as typical absence epilepsy (petit mal). The Lennox-Gastaut syndrome may persist into adult life and is one of the most difficult forms of epilepsy to treat.
The phenomenon of myoclonus was discussed in Chap. 6, where the relationship to seizures was emphasized. Characterized by a brusque, brief, muscular contraction, some myoclonic jerks may be so small as to involve only one muscle or part of a muscle; others are so large as to displace a limb on one or both sides of the body or the entire trunk musculature. Many are brief, lasting 50 to 100 ms; they may occur intermittently and unpredictably or present as a single jerk or a brief salvo.
As mentioned earlier, a series of several small, rhythmic myoclonic jerks may appear with varying frequency as part of atypical absence seizures, and as isolated events in patients with generalized clonic-tonic-clonic or tonic-clonic seizures. As a rule, seizure-associated myoclonus, when occurring in isolation, is relatively benign and usually responds well to medication. In contrast, there are diseases in which myoclonus is progressive in severity or very frequent. These disorders have their onset in childhood and raise the suspicion of entities such as the myoclonus-opsoclonus-ataxia syndrome, lithium or other drug toxicity or, if lasting a few weeks, subacute sclerosing panencephalitis. Chronic progressive polymyoclonus with dementia characterizes the group of juvenile lipidosis, Lafora-type familial myoclonic epilepsy, certain mitochondrial disorders, and other chronic familial degenerative diseases of undefined type (paramyoclonus multiplex of Friedreich) as noted in Table 16-3.
GENE | PROTEIN INVOLVED | |
|---|---|---|
Channelopathies | ||
Sodium channels | ||
Familial generalized seizures with febrile seizures “plus”; see text | SCN1A,B, (GABAA) | Sodium channel subunits; less often, GABA receptor |
Benign familial neonatal convulsions | SCN2A | Sodium channel subunits |
Dravet syndrome (severe myoclonic epilepsy of infancy) | SCN1A | Sodium channel α-subunit |
Potassium channels | ||
Benign infantile epilepsy | KCNQ2,3 | Potassium channel subunits |
Episodic ataxia type 1 with partial epilepsy | KCNA1 | |
Ligand-gated channels | ||
Autosomal dominant nocturnal frontal seizures | CHRNA 2,4 | Nicotinic acetylcholine receptor subunits |
Familial generalized and febrile seizures | GABRG2 | GABAA receptor subunit |
Juvenile myoclonic epilepsy | GABRA1 (CACNB4) | GABAA receptor subunit; less often, calcium channel subunit |
Glucose transporter-1 deficiency | SLC2A1 | GLUT1 (responsive to ketogenic diet) |
Calcium channels | ||
Episodic ataxia type 2 with spike-wave seizures | CACNA1A | Calcium channel subunit |
Malformations of cortical development | ||
Holoprosencephaly, generalized epilepsy | SHH, PTCH, ZIC2, SIX3, TGIF | Sonic hedgehog, SHH receptor, transcription factors |
Schizencephaly, generalized epilepsy | EMX2 | Homeodomain protein |
Tuberous sclerosis, generalized epilepsy | TSC1, 2 | Hamartin, tuberin |
Lissencephaly, generalized epilepsy | LIS1 | Platelet-activating factor acid hydrolase |
Double-cortex syndrome, generalized epilepsy | DCX | Doublecortin |
Heterotopia, focal epilepsy | FLN1 | Filamin1 |
Fukuyama muscular dystrophy, lissencephaly, generalized epilepsy | FCMD | Fukutin |
Walker-Warburg syndrome, generalized epilepsy | POMT1 | O-mannosyl transferase |
Muscle-eye-brain disease, generalized epilepsy | MEB | Glycosyltransferase, PMGnT1 |
Angelman syndrome: myoclonic, tonic-clonic, atonic seizures | UBE3A | Ubiquitin-protein ligase |
Progressive myoclonic epilepsies (PME) | ||
Unverricht-Lundborg disease with PME | EPM1 | Cystatin B |
Lafora body disease with PME | EPM2A | Laforin, protein tyrosine phosphatase |
Myoclonic epilepsy with ragged red fibers | tRNAlys | Mitochondrial lysine tRNA |
Dentatorubro-pallidoluysian atrophy with PME | DRPLA | Atrophin-1 |
Gaucher disease | PSAP | β-Glucocerebrosidase |
Sialidosis type I | NEU1 | Sialidase |
Ceroid lipofuscinosis (CLN) and PME | CLN | CLN2, CLN3, CLN5, CLN6 also cause generalized, atonic and atypical absence seizures |
Mixed seizure types | ||
Lipoid proteinosis and temporal lobe epilepsy | ECM1 | Extracellular matrix protein 1 |
Autosomal dominant lateral temporal lobe epilepsy | LGI1 | Leucine-rich glioma inactivated protein |
CLN8; progressive nonmyoclonic epilepsy with retardation | CLN8 | Membrane protein in endoplasmic reticulum |
Pyridoxine deficiency | ALDH7A1 | Antiquitin (ATQ-1) |
The large number of adult diseases causative of myoclonus and seizure disorders are discussed in Chaps. 33, 37, and 39. Myoclonus as a phenomenon is described in Chap. 6.
This is the most common form of idiopathic generalized epilepsy in older children and young adults. It begins in adolescence, typically around age 15 years, with a range that essentially spans all of the teenage years. The patient comes to attention because of a generalized tonic-clonic seizure, often upon awakening or because of myoclonic jerks in the morning that involve the entire body; sometimes absence seizures are prominent. The family reports that the patient has occasional myoclonic jerks of the arm and upper trunk that is brought out with fatigue, early stages of sleep, or alcohol ingestion. A few patients in our experience have had only the myoclonic phenomena and rare absence or tonic-clonic seizures that persisted unnoticed for years. The EEG shows characteristic bursts of 4- to 6-Hz irregular polyspike activity. A linkage has been established to chromosome 6 in some cases of this illness, and in some other forms of juvenile-onset epilepsy, and several are the result of mutations in ion channel genes.
The disorder does not impair intelligence and tends not to be progressive, for which reason it has been called “benign”, but a proclivity to infrequent seizures usually continues throughout life. A report by Bakan and colleagues has indicated that, over an average of two decades, the majority of patients have long seizure-free periods and a great reduction in myoclonic seizures. Valproic acid in particular and some other antiepileptic drugs are highly effective in eliminating the seizures and myoclonus but they should be continued indefinitely as discontinuation of medication is associated with a high rate of relapse. Owing to the potential teratogenicity of valproate, women of childbearing age are often given levitiracetam or lamotrigine, acknowledging that they may not be as effective as the first choice of drug.
Focal Seizures
As indicated earlier, the International Classification divides all seizures into two types—generalized, in which the clinical and EEG manifestations indicate bilateral and diffuse cerebral cortical involvement from the onset, and focal, in which the seizure is often the product of a demonstrable focal lesion or EEG abnormality in some part of the cerebral cortex. Partial seizures vary with the locale of the lesion and are conventionally qualified based their specific clinical characteristics and on whether consciousness is retained or impaired. Focal seizures with sensory or motor features at the onset most often arise from foci in the sensorimotor cortex. Those with impairment of consciousness, which occurs in many forms, most often have their focus in the limbic and autonomic areas or in the temporal lobe, but a frontal localization is also known. Table 16-2, on a previous page, lists the common sites of the lesions and the types of seizures to which they give rise.
Relatively few focal seizures can be localized precisely from clinical data alone. However, when combined with scalp and intracranial EEG recording and MRI, the localization is reasonably accurate.
Focal or partial motor seizures are attributable to a discharging lesion of the frontal lobe. The most common type, originating in the supplementary motor area, takes the form of a turning movement of the head and eyes to the side opposite the irritative focus, often associated with a tonic extension of limbs, also on the side contralateral to the affected hemisphere. This may constitute the entire seizure, or it may be followed by generalized clonic movements. The extension of the limbs may occur just before or simultaneously with loss of consciousness but a lesion in one frontal lobe may give rise to a major generalized convulsion without an initial turning of the head and eyes. It has been postulated that in both types of seizures, the one with and the one without turning movements, there is an immediate spread of the discharge from the frontal lobe to integrating centers in the thalamic or high midbrain reticular formation, accounting for the loss of consciousness.
The frontal lobe, being so large, can give rise to numerous forms of seizure. In addition to the typical Jacksonian type described above, there are adversive, speech arrest, frontal, absence types, and a number of unusual disorders related to discharges from the supplementary motor area including hyperkinetic and postural tonic varieties. In practice, it is often difficult to distinguish such seizures from parasomnic (sleep related) events (see Chap. 18).
Jacksonian seizures begin with forceful, sustained deviation of the head and eyes, and sometimes of the entire body, are referred to as versive or adversive. Because the turning movements are usually to the side opposite the irritative focus (sometimes to the same side), contraversive and ipsiversive, respectively, might be preferable terms. Nonforceful, unsustained, or seemingly random lateral head movements during the ictus do not have localizing value. The same is true for the head and eye turning that occurs at the end of the generalized tonic-clonic phase of seizures (Wylie et al). Contraversive deviation of only the head and eyes can be induced most consistently by electrical stimulation of the superolateral frontal region (area 8), just anterior to area 6 (see Figs. 22-1 and 22-2). In seizures of temporal lobe origin, early in the seizure, there may be head turning ipsilaterally followed by forceful, contraversive head (and body) turning. These head and body movements, if they occur, are preceded by quiet staring or automatisms.
The Jacksonian motor seizure may also begin with a tonic contraction of the fingers of one hand, the face on one side, or the muscles of one foot. This transforms into clonic movements in these parts in a fashion analogous to that in a generalized clonic-tonic-clonic convulsion. Sometimes a series of clonic movements of increasing frequency build up to a tonic contraction. The movements spread (“march”) from the part first affected to other muscles on the same side of the body. In this typical Jacksonian form, the seizure spreads from the hand, up the arm, to the face, and down the leg; or if the first movement is in the foot, the seizure marches up the leg, down the arm, and to the face, usually in a matter of 20 to 30 s. Interestingly, spontaneously occurring focal motor seizures, e.g., those beginning in the toes or fingers, may sometimes be arrested (inhibited) by applying a ligature above the affected part or, in the case of focal sensory seizures, by applying a vigorous sensory stimulus ahead of the advancing sensory aura. Rarely, the first muscular contraction is in the abdomen, thorax, or neck. In some cases, the one-sided seizure activity is followed by turning of the head and eyes to the convulsing side, occasionally to the opposite side, and then by a generalized seizure with loss of consciousness. Consciousness is not lost if the sensorimotor symptoms remain confined to one side.
Following convulsions that have a prominent focal motor signature, there may be a transient paralysis of the affected limbs. This “Todd’s paralysis” persists for minutes or at times for hours after the seizure, usually in proportion to the duration of the convulsion. Continued focal paralysis beyond this time usually indicates the presence of a focal brain lesion as the underlying cause of the seizure or persisting seizures in a nonconvulsive form. A similar Todd phenomenon is found in cases of focal epilepsy that involve the language, somesthetic, or visual areas; here the persistent deficit corresponds to the region of brain affected.
The high incidence of focal motor epilepsy that originates with movements in the face, hands, and toes is probably related to the disproportionately large cortical representation of these parts. The disease process or focus of excitation is usually in or near the rolandic (motor) cortex, i.e., area 4 of Brodmann (Figs. 3-3 and 22-2); in some cases, and especially if there is a sensory accompaniment, it has been found in the postrolandic convolution. Lesions confined to the motor cortex are reported to assume the form of clonic contractions, and those confined to the premotor cortex (area 6), tonic contractions of the contralateral arm, face, neck, or all of one side of the body. Tonic elevation and extension of the contralateral arm (“fencing posture”) and choreoathetotic and dystonic postures have been associated with high medial frontal lesions (area 8 and supplementary motor cortex), as have complex, bizarre, and flailing movements of a contralateral limb, but this always raises the suspicion of hysterical seizure. Perspiration and piloerection occur occasionally in parts of the body involved in a focal motor seizure, suggesting that these autonomic functions have a cortical representation in or adjacent to the rolandic area. Focal motor and Jacksonian seizures have essentially the same localizing significance.
Seizure discharges arising from the cortical language areas may give rise to a brief aphasic disturbance (ictal aphasia) and ejaculation of a word or, more frequently, a vocal arrest. Ictal aphasia is usually succeeded by other focal or generalized seizure activity but may occur in isolation, without loss of consciousness, in which case it can later be described by the patient. Postictal aphasia is more common than ictal aphasia, which typically takes the form of complete speech arrest. Verbalization at the onset of a seizure has no consistent lateralizing significance and, paradoxically, is usually associated with an origin in the nondominant hemisphere. These disturbances should be distinguished from the stereotyped repetition of words or phrases or the garbled speech that characterizes some complex partial seizures or the postictal confusional state and, of course, Wernicke aphasia.
Somatosensory seizures, either focal or “marching” to other parts of the body on one side, are nearly always indicative of a focus in or near the postrolandic convolution of the opposite cerebral hemisphere. Penfield and Kristiansen found the seizure focus in the postcentral or precentral convolution in 49 of 55 such cases. The sensory disorder is usually described as numbness, tingling, or a “pins-and-needles” feeling and occasionally as a sensation of crawling (formication), electricity, or movement of the part. Pain and thermal sensations may occur but are exceedingly rare. In the majority of cases, the onset of the sensory seizure is in the lips, fingers, or toes, and the spread to adjacent parts of the body follows a pattern determined by sensory arrangements in the postcentral (postrolandic) convolution of the parietal lobe. If the sensory symptoms are localized to the head, the focus is in or adjacent to the lowest part of the convolution, near the sylvian fissure; if the symptoms are in the leg or foot, the upper part of the convolution, near the superior sagittal sinus or on the medial surface of the hemisphere, is involved.
Olfactory hallucinations, perhaps the most important of the sensory seizures because they signify a particular localization, are associated with disease of the inferior and medial parts of the temporal lobe, usually in the region of the parahippocampal convolution or the uncus (hence Jackson’s term uncinate seizures [see also Chap. 12]). Usually the perceived odor is exteriorized, i.e., projected to someplace in the environment, and is described as disagreeable or foul, though otherwise unidentifiable. Gustatory hallucinations also have been recorded in proven cases of temporal lobe disease and less often with lesions of the insula and parietal operculum; salivation and a sensation of thirst may be associated. Electrical stimulation in the depths of the sylvian fissure, extending into the insular region, has produced peculiar sensations of taste.
Visual seizures are relatively rare but also have localizing significance. Lesions in or near the striate cortex of the occipital lobe usually produce elemental visual sensations of darkness or sparks and flashes of light, which may be stationary or moving and colorless or colored. According to Gowers, red is the most frequently reported color, followed by blue, green, and yellow. These images may be referred to the visual field on the side opposite of the lesion or may appear straight ahead. If they occur on one side of the visual field, patients perceive that only one eye is affected (the one opposite the lesion), probably because most persons are aware of only the temporal half of a homonymous field defect. Curiously, a seizure arising in one occipital lobe may cause momentary blindness in both fields. It has been noted that lesions on the lateral surface of the occipital lobe (Brodmann areas 18 and 19) are likely to cause a sensation of twinkling or pulsating lights. More complex or formed visual hallucinations are usually caused by a focus in the posterior part of the temporal lobe, near its junction with the occipital lobe, and may be associated with auditory hallucinations. The localizing value of visual auras has been confirmed by Bien and colleagues in a group of 20 surgically treated patients with intractable seizures. They found that elementary visual hallucinations and visual loss were typical of occipital lobe epilepsy but could also occur with seizure foci in the anteromedial temporal and occipitotemporal regions.
Auditory hallucinations are infrequent as an initial manifestation of a seizure and usually represent a psychotic disorder or one of several more benign conditions. Occasionally, a patient with a focus in one superior temporal convolution will report a buzzing or roaring in the ears. A human voice, sometimes repeating unrecognizable words, or the sound of music has been noted a few times with lesions in the more posterior part of one temporal lobe. Some people with epilepsy and a strong family history of seizures with auditory auras, may have normal imaging but turn out to have mutations in the LGI1 gene.
Vertiginous sensations of a type suggesting a vestibular origin may on rare occasions be the first symptom of a seizure. The lesion is usually located in the superoposterior temporal region or the junction between parietal and temporal lobes. In one of the cases reported by Penfield and Jasper, a sensation of vertigo was evoked by stimulating the cortex at the junction of the parietal and occipital lobes. Occasionally with a temporal focus, the vertigo is followed by an auditory sensation. Giddiness, or light-headedness, is a frequent prelude to a seizure, but this symptom, as discussed in Chap. 15, has so many different connotations that it is of little diagnostic value.
Vague and often indefinable visceral sensations arising in the thorax, epigastrium, and abdomen are among the most frequent of auras, as already indicated. Most often they have a temporal lobe origin, although in several such cases the seizure discharge has been localized to the upper bank of the sylvian fissure, in the upper or middle frontal gyrus, or in the medial frontal area near the cingulate gyrus. Palpitation and acceleration of the heart rate at the beginning of the attack have also been related mainly to a temporal lobe focus.
Focal Seizures Characterized by Altered Awareness or Responsiveness (Formerly Termed Complex Partial Seizures, Psychomotor Seizures, Temporal Lobe Seizures)
These differ from the major generalized and absence seizures discussed above in that (1) they signify a focal onset in the temporal lobe as reflected by an aura that may be a hallucination or perceptual illusion, and (2) instead of a complete loss of control of thought and action, there is a period of altered behavior and consciousness, for which the patient is later found to be amnesic.
Although it is difficult to enumerate all the psychic experiences that may occur during these types of seizures, they may be categorized into a somewhat arbitrary hierarchy of illusions, hallucinations, depersonalization states, and affective experiences. Sensory illusions, or distortions of ongoing perceptions, are the most common. Objects or persons in the environment may shrink or recede into the distance, or they may enlarge (micropsia and macropsia), or perseverate as the head is moved (palinopsia). Tilting of the visual environment has been reported. Hallucinations are most often visual or auditory, consisting of formed or unformed visual images, sounds, and voices; less frequently, they may be olfactory (usually unpleasant, unidentifiable sensations of smell), gustatory, or vertiginous. Among the altered psychic states are a feeling of intense perception of familiarity in an unfamiliar circumstance or place (déjà vu) or, conversely, of strangeness or unfamiliarity (jamais vu) in a previously known place or circumstance. There may be the experience of autoscopy, a type of depersonalization, or dream-like state in which the patient views himself as an external observer. Fragments of certain old memories or scenes may insert themselves into the patient’s mind and recur with striking clarity, or there may be an abrupt interruption of memory. (See Gloor for a more detailed description of the experiential phenomena of temporal lobe epilepsy.) Associated epigastric and abdominal sensations have been alluded to above and likely have their origin in autonomic and limbic structures.
Emotional experiences as a result of seizure, while less common, may be dramatic—fear, sadness, loneliness, anger, happiness, and sexual excitement have all been recorded. Fear and anxiety are the most common affective experiences, while occasionally the patient describes a feeling of rage or intense anger as part of a complex partial seizure. Ictal fear has no apparent connection to objective experience and is generally not related to the situation in which the patient finds himself during the seizure.
Each of these subjective psychic states may constitute the entire seizure or some combination may occur and immediately precedes a period of altered awareness. These “auras” represent electrical seizures as already mentioned and have the same localizing significance as motor convulsions do for the frontal cortex.
The motor components of a focal temporal lobe or limbic seizure, if they occur, arise during the later phase of the seizure and take the form of automatisms such as lip-smacking, chewing or swallowing movements, salivation, fumbling of the hands, or shuffling of the feet. Patients may walk around in a daze or act inappropriately (undressing in public, speaking incoherently, etc.). Certain complex acts that were initiated before the loss of consciousness—such as walking, chewing food, turning the pages of a book, or even driving—may continue. However, when asked a specific question or given a command, the patients are obviously out of contact with their surroundings. There may be no response at all, or the patient may look toward the examiner in a perplexed way or utter a few stereotyped phrases. The patient may walk repetitively in small circles (volvular epilepsy), run (epilepsia procursiva), or simply wander aimlessly, either as an ictal or postictal phenomenon (poriomania). These forms of seizure, according to some epileptologists, are actually more common with frontal lobe than with temporal lobe foci of origin.
In a very small number of patients with temporal lobe seizures (7 of 123 patients studied by Ebner et al), some degree of responsiveness (to simple questions and motor commands) is preserved in the presence of prominent automatisms such as lip-smacking and swallowing. Interestingly, in this small group of partially responsive patients, the seizures originate in the right temporal lobe.
The patient, in a confused and irritable state, may resist or strike out at the examiner. These types of behaviors, which occur in a limited number of patients with temporal lobe or frontal seizures, usually take the form of nondirected oppositional resistance to restraint. These behaviors manifest during a period of automatic behavior (so called because the patient presumably acts like an automaton) or, more often, in the postictal period. Unprovoked assault or outbursts of intense rage or blind fury are very unusual; Currie and associates found such outbursts in only 16 of 666 patients (2.4 percent) with temporal lobe epilepsy. Penfield once commented that he had never observed a rage state as a result of temporal lobe stimulation. It is exceedingly unlikely that an organized violent act requiring several sequential steps in its performance, such as obtaining a weapon and using it in a directed manner, could represent a temporal lobe seizure.
Rarely, laughter may be the most striking feature of a seizure (gelastic epilepsy). A particular combination of gelastic seizures and precocious puberty has been traced to a hamartoma of the hypothalamus. Crying, on the other hand, is very infrequent as a component of seizure and usually indicates a psychogenically induced episode.
Dystonic stiffness of the arm and leg contralateral to the seizure focus is found to be an accompaniment of temporal lobe seizures (more often this is from the supplementary motor of the frontal than the temporal lobes).
The patient with temporal lobe seizures may exhibit only one of the foregoing manifestations of seizure activity or various combinations. In a series of 414 patients studied by Lennox, 43 percent displayed some of the motor changes; 32 percent, automatic behavior; and 25 percent, alterations in psychic function. Because of the frequent concurrence of these symptom complexes, he referred to them as the psycho-motor triad. Probably the clinical pattern varies with the precise locality of the lesion and the direction and extent of spread of the electrical discharge.
After the attack, the patient usually has no memory or only fragments of recall for what was said or done. Any type of complex partial seizures may proceed to other forms of secondary generalized seizures. The tendency to generalization holds true for all types of partial or focal epilepsy.
Temporal lobe seizures are not peculiar to any period of life, but they do show an increased incidence in adolescence and the adult years and have an uncertain relationship to febrile seizures. The topic of febrile seizures is broader than this association suggests; it is taken up in a later section of the chapter. Neonatal convulsions, head trauma, and various other non-progressive perinatal neurologic disorders are other antecedents that place a child at risk of developing complex partial seizures (Rocca et al). Two-thirds of patients with temporal lobe seizures also have generalized tonic-clonic seizures or have had them in early childhood, and it has been theorized that the generalized seizures may have led to secondary excitotoxic damage to the hippocampal portions of the temporal lobes. In the latter cases, carefully performed and quantitated MRI in the coronal plane may disclose a loss of volume in the hippocampi and adjacent gyri on one or both sides—i.e., medial temporal sclerosis (Fig. 16-4).
Figure 16-4.
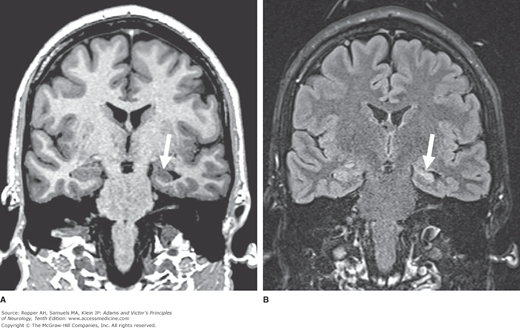
Medial temporal sclerosis. A. T1-weighted MRI in the coronal plane, showing reduced volume of the left hippocampus (shown by arrow) and secondary enlargement of the adjacent temporal horn of the lateral ventricle. B. Coronal T2-FLAIR image showing abnormal hyperintense signal within the left hippocampus (shown by arrow).
Temporal lobe seizures are highly variable in duration. Behavioral automatisms rarely last longer than a minute or two, although postictal confusion and amnesia may persist for a considerably longer time. Some consist only of a momentary change in facial expression and a blank spell, resembling an absence. Almost always, however, temporal lobe events are characterized by distinct ictal and postictal phases, whereas patients with absence attacks usually have an instantaneous return of full consciousness following the ictus.
Postictal behavior after temporal lobe seizures is often accompanied by widespread or focal slowing in the EEG. Prolonged disorientation for time and place suggests a right-sided source. Automatisms in the postictal period have no lateralizing connotation (Devinsky et al). However, postictal posturing and paresis of an arm (Todd’s paralysis) or an aphasic difficulty are helpful in determining the side of the lesion (Cascino). Postictal nose wiping, which is reported on video recording to occur in half of patients with temporal lobe seizures, is carried out by the hand ipsilateral to the seizure focus according to Leutzmezer and colleagues, but we are in no position to confirm this.
Rarely, recurrent attacks of transient amnesia are the only manifestations of temporal lobe epilepsy, although it is unclear whether the amnesia in such patients represents an ictal or postictal phenomenon. These attacks of pure amnesia have been referred to as transient epileptic amnesia (TEA; Palmini et al; Zeman et al). If the patient functions at a fairly high level during the attack, as may happen, there is a resemblance to transient global amnesia (described in Chap. 20). However, in contrast to transient global amnesia, the relative brevity and frequency of the amnesic spells, their tendency to occur on awakening, the impaired performance on complex cognitive tasks, and the absence of repetitive stereotyped questions help to make the distinction.
Some comments are in order concerning the issues of behavioral and psychiatric disorders in patients who have seizures. Data as to prevalence of these disorders have been derived mainly from studies of selected groups of patients attending university hospitals and other specialty clinics that tend to treat the most difficult and complicated cases. In one such study (Victoroff), approximately one-third of epileptic patients had a history of major depressive illness, and an equal number had symptoms of anxiety disorder; psychotic symptoms were found in 10 percent. Similar figures, also from a university-based epilepsy center, have been reported by Blumer et al. It must be emphasized that these remarkably high rates of psychiatric morbidity do not reflect the prevalence in the entire population of patients with epilepsy. Epidemiologic studies provide only limited evidence of an association with psychosis in the overall population of epileptics (see Trimble and Schmitz and the review by Trimble for a critical discussion of this subject). Furthermore, it should be borne in mind that many chronic medical conditions are associated with psychiatric reactions. On the other hand, the unpredictability and stigma of the epileptic disorders may contribute to depression and anxiety.
The postictal state in patients with temporal lobe epilepsy rarely incorporates a protracted paranoid-delusional or amnesic psychosis lasting for days or weeks. The EEG during this period may show no seizure discharge, although this does not exclude repeated seizures in temporal lobe structures that are remote from the recording electrodes. This disorder, virtually indistinguishable from psychosis, may also present in the interictal period.
It had been observed that some patients with temporal lobe seizures may exhibit a number of personal peculiarities. It was stated that they are slow and rigid in their thinking, verbose, circumstantial and tedious in conversation, inclined to mysticism, and preoccupied with rather naive religious and philosophical ideas. Obsessionalism, humorless sobriety, emotionality (mood swings, sadness, and anger), and a tendency to paranoia are other frequently described traits. Diminished sexual interest and potency in men and menstrual problems in women, not readily attributable to antiepileptic drugs, are common among patients with complex partial seizures of temporal lobe origin. Geschwind proposed that a triad of behavioral abnormalities—hyposexuality, hypergraphia, and hyperreligiosity—constitutes a characteristic syndrome.
Bear and Fedio suggested that certain personality traits were more common with right temporal lesions, and that anger, paranoia, and cosmologic or religious conceptualizing are more characteristic of left temporal lesions. However, Rodin and Schmaltz found no features that would distinguish foci on either side and they found no behavioral changes that would distinguish patients with temporal lobe epilepsy from other groups of epileptics. The problem of personality disturbances in epilepsy has not been clarified and modern clinicians no longer identify these traits as parts so the epileptic syndrome, having in the past been imputed to these patients by societal and medical biases (see review by Trimble).
In the past few decades, sudden death has been emphasized as an underappreciated problem in the epileptic population. Certainly, the mortality in individuals with epilepsy is increased ostensibly from accidents, suicide, and the underlying cause of seizures. However, the main contributor to the increased mortality rate in healthy people with epilepsy is unexpected death outside of circumstances such as drowning, trauma from a fall, myocardial infarction, and automobile accidents during the seizure. It is to this group that the acronym “SUDEP” has been applied. Surprisingly, unexpected death is predominantly an issue of adulthood more than of childhood. The rate of unexpected death increases with the duration and severity of epilepsy and several population studies suggest that the rate may be as high as 9% lifetime incidence but others cite a much lower figure. Most patients affected have a history of generalized tonic-clonic seizures and die in bed. In children, those with treatment resistant epilepsy, developmental delay and several syndromes such as tuberous sclerosis are at particular risk.
Several factors have emerged as risks from population-based and cohort case controlled studies; the postictal period immediately after a tonic clonic seizure, increasing seizure frequency (including three generalized seizures in the preceding year), lack of successful treatment (i.e., patients not in remission as documented in a 40 year follow up of childhood epilepsy by Sillanpää and Shinnar), or subtherapeutic levels of antiepileptic drugs, the period of early adulthood, long-standing epilepsy, and mental retardation.
Most instances of SUDEP occur when the patient is unattended or during sleep. Although respiratory difficulty and cardiac changes including asystole and ventricular arrhythmias are known to occur during and immediately after seizures, none of these has been a consistent factor and usually, the precise mechanism of death has been difficult to determine. A postictal “shutdown” of brainstem activity resulting in hypercapnia or hypoxemia has been suggested.
One approach to preventing sudden death is adequate treatment with antiepileptic drugs. The risk of sudden death is as high as 20 times greater for untreated patients. Some specialists in the field of epilepsy have suggested that an open conversation be undertaken about the problem with patients and their families. More often, neurologists raise the issue only in high risk patients or when specifically asked. A review of this subject has been provided by Devinsky.
Special Epileptic Syndromes
There remain to be considered several epileptic syndromes and other seizure states that cannot be readily classified with the usual types of generalized or partial seizures.
This common focal motor epilepsy is unique among the focal epilepsies of childhood in that it is self-limiting despite a very abnormal EEG pattern. It is usually transmitted in families as an autosomal dominant trait and begins between 5 and 9 years of age. It typically announces itself by a nocturnal tonic-clonic seizure with focal onset. Thereafter, the seizures take the form of clonic contractions of one side of the face, less often of one arm or leg, and the interictal EEG shows high-voltage spikes in the contralateral lower rolandic or centrotemporal area. Seizures are readily controlled by a single anticonvulsant drug and gradually disappear during adolescence. The relation of this syndrome to developmental dyslexia is unsettled.
A similar type of epilepsy, usually benign in the sense that there is no intellectual deterioration and the seizures often cease in adolescence, has been associated with spike activity over the occipital lobes as identified by Panayiotopoulos. Visual hallucinations, while not invariable, are the most common clinical feature, according to the review by Taylor and colleagues; sensations of movements of the eyes, tinnitus, or vertigo are also reported in cases of occipital epilepsy. These authors point out symptomatic causes of the syndrome, mainly cortical heterotopias. In both of these types of childhood epilepsy the observation that spikes are greatly accentuated by sleep is a useful diagnostic sign.
This term is applied to a special and particularly dramatic form of epilepsy of infancy and early childhood. West, in the mid-nineteenth century, described the condition in his son in great detail. This disorder, which in most cases appears during the first year of life, is characterized by recurrent, single or brief episodes of gross flexion movements of the trunk and limbs and, less frequently, by extension movements (hence the alternative terms infantile spasms or salaam or jackknife seizures). Most but not all patients with this disorder show major EEG abnormalities consisting of continuous multifocal spikes and slow waves of large amplitude. However, this pattern, named by Gibbs and Gibbs as hypsarrhythmia (“mountainous” dysrhythmia), is not specific for infantile spasms, being frequently associated with other developmental or acquired abnormalities of the brain. As the child matures, the seizures diminish and usually disappear by the fourth to fifth year. If MRI and CT scans are more or less normal, the usual pathologic findings according to Jellinger are cortical dysgeneses. Both the seizures and the EEG abnormalities may respond dramatically to treatment with adrenocorticotropic hormone (ACTH), corticosteroids, or the benzodiazepine drugs, of which clonazepam is probably the most widely used. A type of West syndrome that is caused by tuberous sclerosis also responds dramatically to gamma-aminobutyric acid (GABA)-inhibiting drugs such as vigabatrin, as noted below. However, most patients, even those who were apparently normal when the seizures appeared, are left mentally impaired. Infantile spasms may later progress to the Lennox-Gastaut syndrome, a seizure disorder of early childhood of graver prognosis as discussed in a previous section.
The well-known uncomplicated febrile seizure, specific to infants and children between 6 months and 5 years of age (peak incidence ages 9 to 20 months) and with a strong inherited tendency, is generally regarded as a benign condition. It usually takes the form of a single, generalized motor seizure occurring as the patient’s core temperature rises or reaches its peak. Seldom does the seizure last longer than a few minutes and by the time an EEG can be obtained, there is no abnormality and recovery is complete. The temperature is usually above 38°C (100.4°F). Any viral or bacterial illness, or, rarely, an immunization, may be the precipitant of the fever; herpesvirus 6 is one of the common precipitants, probably because of its tendency to cause high fever. Prophylactic antiepileptic drugs have not been found to be helpful in preventing febrile seizures. Except for a presumed genetic relationship with benign epilepsy of childhood (Luders et al), which in itself is transient in nature, these patients’ risk of developing epilepsy in later life is only slightly greater than that of the general population. In some families, such as those studied by Nabbout and colleagues, febrile seizures alone, without generalized epilepsy, have been associated with a particular gene by linkage analysis. Presumably, when the gene products are identified, some insight into the nature of defects that lower the seizure threshold will be forthcoming.
This benign type of febrile seizure should not be confused with more serious illnesses in which a febrile acute encephalitic or encephalopathic state causes focal or prolonged seizures, generalized or focal EEG abnormalities, and repeated episodes of febrile convulsions during a febrile illness (complicated febrile seizures). In these cases, these seizures may recur not only with infections but also at other times. When patients with both types are combined together under the rubric of febrile convulsions, it is not surprising that a high percentage are complicated by later atypical petit mal, atonic, and astatic spells followed by tonic seizures, mental retardation, and partial complex epilepsy. In a study of 67 patients with medial temporal lobe epilepsy by French and colleagues, 70 percent had a history of complicated febrile seizures during the first 5 years of life, although many did not again develop seizures until their teens. Bacterial meningitis was an important risk factor; head and birth trauma were less-common factors. Epidemiologic studies have substantiated this clinical point of view. Annegers and colleagues observed a cohort of 687 children for an average of 18 years after their initial febrile convulsion. Overall, these children had a five-fold excess of unprovoked seizures in later life. Among the children with simple febrile convulsions, the risk was only 2.4 percent. By contrast, children with what Annegers and colleagues called complex febrile convulsions (focal, prolonged, or repeated episodes of febrile seizures) had a greatly increased risk—8, 17, or 49 percent, depending on the association of one, two, or three of the complicating features.
It has been appreciated for a long time that seizures can be evoked in certain individuals by a discrete physiologic or psychologic stimulus. The term reflex epilepsy is reserved for this small subgroup. Forster classified these seizures in accordance with their evocative stimuli into five types: (1) visual—flickering light, visual patterns, and specific colors (especially red), leading to rapid blinking or eye closure; (2) auditory—sudden unexpected noise (startle), specific sounds, musical themes, and voices; (3) somatosensory—either a brisk unexpected tap or sudden movement after sitting or lying still, or a prolonged tactile or thermal stimulus to a certain part of the body; (4) writing or reading of words or numbers; and (5) eating.
Visually induced seizures are by far the most common type. The seizures are usually myoclonic but may be generalized and triggered by the photic stimulation of television or an EEG examination or by the photic or pattern stimulation of video games. In other types of reflex epilepsy, the evoked seizure may be focal (beginning often in the part of the body that was stimulated) or generalized and may take the form of one or a series of myoclonic jerks or of an absence or tonic-clonic seizure. Seizures induced by reading, voices, or eating are most often of the complex partial type; seizures induced by music are usually myoclonic, simple, or complex partial. A few such instances of reflex epilepsy have been caused by focal cerebral disease, particularly occipital lesions.
Clonazepam, valproate, carbamazepine, and phenytoin (as well as many of the new antiepileptic drugs) are all effective in controlling individual instances of reflex epilepsy. Some patients learn to avert the seizure by undertaking a mental task, e.g., thinking about some distracting subject, counting, etc., or by initiating some type of physical activity. Forster has demonstrated that in certain types of reflex epilepsy, the repeated presentation of the stimulus may eventually render the trigger innocuous but this requires a great deal of time and reinforcement, which limits its therapeutic value.
This is a special type of focal motor epilepsy characterized by persistent rhythmic clonic movements of one muscle group—usually of the face, arm, or leg—which are repeated at fairly regular intervals every few seconds and continue for hours, days, weeks, or months without spreading to other parts of the body. Thus epilepsia partialis continua is, in effect, a highly restricted and very persistent focal motor status epilepticus. The distal muscles of the leg and arm, especially the flexors of the hand and fingers, are affected more frequently than the proximal ones. In the face, the recurrent contractions involve either the corner of the mouth or one or both eyelids. Occasionally, isolated muscles of the neck or trunk are affected on one side. The clonic activity may be accentuated by active or passive movement of the involved muscles and may be reduced in severity but not abolished during sleep.
First described by Kozhevnikov in patients with Russian spring-summer encephalitis, these ongoing partial seizures may be induced by a variety of acute or chronic cerebral lesions. In some cases the underlying disease is not apparent (this has applied to most of the cases in our experience), and the clonic movements may be mistaken for some type of slow tremor or extrapyramidal movement disorder. Most patients with epilepsia partialis continua show focal EEG abnormalities, either repetitive slow-wave abnormalities or sharp waves or spikes over the central areas of the contralateral hemisphere. In some cases, the spike activity can be related precisely in location and time to the clonic movements (Thomas et al). In the series collected by Obeso and colleagues, there were various combinations of epilepsia partialis continua and cutaneous reflex myoclonus (cortical myoclonus occurring only in response to a variety of afferent stimuli).
As would be expected, a wide range of causative lesions has been implicated—developmental anomalies, encephalitis, demyelinative diseases, tumors, metabolic abnormalities, particularly hyperosmolarity, and degenerative diseases; but in many instances, as already emphasized, the underlying cause is not found even after extensive investigation. Epilepsia partialis continua is particularly common in patients with the rare condition, Rasmussen encephalitis (see further on).
Whether cortical or subcortical mechanisms are responsible for epilepsia partialis continua is an unresolved question. The electrophysiologic evidence adduced by Thomas and colleagues favors a cortical origin; the pathologic evidence is less definite. In each of eight cases in which the brain was examined postmortem, they found some degree of involvement of the motor cortex or adjacent cortical area contralateral to the affected limbs. However, all but one of these patients also had some involvement of deeper structures on the same side as the cortical lesion, on the opposite side, or on both sides.
In rare cases, a lesion, usually identified by MRI and confirmed by biopsy, and in some cases by special autoantibodies, takes the form of a chronic focal encephalitis. In 1958, Rasmussen described three children in whom the clinical problem consisted of intractable focal epilepsy in association with a progressive hemiparesis. The cerebral cortex disclosed a mild meningeal infiltration of inflammatory cells and an encephalitic process marked by neuronal destruction, gliosis, neuronophagia, some degree of tissue necrosis, and perivascular cuffing. Many additional cases were soon uncovered and Rasmussen was able to summarize the natural history of 48 personally observed patients (see the often cited monograph by Andermann).
An expanded view of the syndrome has added several interesting features. The affected children are typically ages 3 to 15 years, more girls than boys. Half of them have epilepsia partialis continua. The progression of the disease leads to hemiplegia or other deficits and focal brain atrophy in most cases. The neuropathology of five fully examined cases has revealed extensive destruction of the cortex and white matter with intense gliosis and lingering inflammatory reactions.
The CSF shows a pleocytosis and sometimes oligoclonal bands but these are not uniform findings. Focal cortical and subcortical lesions are usually visualized by MRI and are bilateral in some cases. The finding of antibodies to glutamate receptors (GluR3) in a proportion of patients with Rasmussen encephalitis has raised interest in an immune causation (see review by Antel and Rasmussen
Related posts:
 Chapter 6. Tremor, Myoclonus, Focal Dystonias, and Tics
Chapter 18. Faintness and Syncope
Chapter 21. Dementia, the Amnesic Syndrome, and the Neurology of Intelligence and Memory
Chapter 26. Disorders of the Autonomic Nervous System, Respiration, and Swallowing
Chapter 42. Alcohol and Alcoholism
Chapter 52. Depression and Bipolar Disease
Chapter 6. Tremor, Myoclonus, Focal Dystonias, and Tics
Chapter 18. Faintness and Syncope
Chapter 21. Dementia, the Amnesic Syndrome, and the Neurology of Intelligence and Memory
Chapter 26. Disorders of the Autonomic Nervous System, Respiration, and Swallowing
Chapter 42. Alcohol and Alcoholism
Chapter 52. Depression and Bipolar Disease
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


