Multiple Sclerosis and Other Inflammatory Demyelinating Diseases: Introduction
It has long been the practice to set apart a group of diseases of the brain and spinal cord in which destruction of myelin, termed demyelination, is a prominent feature. To define these diseases precisely is difficult, for the simple reason that there is probably no disease in which myelin destruction is the exclusive pathologic change. The idea of a demyelinating disease is an abstraction that serves primarily to focus attention on one of the more striking and distinctive features of one group of pathologic processes. Another unifying feature of most of these processes is the participation of an inflammatory reaction in proximity to demyelination. The most common and important inflammatory demyelinating disease is multiple sclerosis (MS).
The generally accepted pathologic criteria of a demyelinating disease are (1) destruction of the myelin sheaths of nerve fibers with relative sparing of the other elements of nervous tissue, i.e., of axons, nerve cells, and supporting structures, which are less affected; (2) infiltration of inflammatory cells, particularly in a perivenous distribution; (3) lesions that are primarily in white matter, either in multiple small disseminated foci or in larger foci spreading from one or more centers. In most of the demyelinating diseases, it has been known since the early descriptions that there is some degree of neuronal and axonal degeneration, but it is the preferential effect on myelin that defines this group of disorder. In the language of neurology, therefore, the term demyelination has acquired a special meaning.
A broad classification of the inflammatory demyelinating diseases is given in Table 36-1. Like all classifications that are not based on etiology, this one has its limitations. For example, in some of the diseases here classified as demyelinating, notably, in necrotizing hemorrhagic leukoencephalitis and even in some cases of multiple sclerosis, the inflammatory process may be sufficiently intense so much so that there is destruction of all tissue in a region including vessels and axons.
|
In contrast, a number of diseases in which demyelination is a prominent feature are considered part of this category, as mentioned earlier. In some cases of anoxic encephalopathy, for example, the myelin sheaths of the radiating nerve fibers in the deep layers of the cerebral cortex or in ill-defined patches in the convolutional and central white matter are destroyed, while most of the axis cylinders are spared. A relatively selective degeneration of myelin may also occur in some small ischemic foci as a result of vascular occlusion or in larger confluent areas, as is the case in Binswanger disease (see Chap. 34). In subacute combined degeneration (SCD) of the spinal cord associated with pernicious anemia and in tropical spastic paraparesis (TSP), a demyelinating spinal cord disease, myelin may be affected earlier and to a greater extent than axons. The same is true of progressive multifocal leukoencephalopathy (PML), osmotic demyelination (also known as central pontine myelinolysis), and Marchiafava-Bignami disease. Some of these disorders and several others are not classified as demyelinating because the effects of the process are not primarily on myelin; furthermore, they are not based on an inflammatory component. In addition, for reasons that will become clear in subsequent discussion, the chronic progressive leukodystrophies of childhood and adolescence (e.g., globoid body, metachromatic, and adrenal leukodystrophies), although clearly diseases of myelin, are set apart and called dysmyelinating because of their unique genetic and morphologic features, and are discussed in Chap. 37. Occupying an uncertain place in this nosology are demyelinating lesions associated with connective tissue diseases or with autoantibodies directed against DNA or phospholipids. The central nervous system (CNS) lesions may be multiple and cannot easily be distinguished by imaging features from multiple sclerosis. But, as noted further on, their nature is uncertain, while some are clearly caused by a vasculopathy.
Multiple Sclerosis
Multiple sclerosis, called MS by most physicians, was referred to by the British as disseminated sclerosis and by the French as sclérose en plaques. The broad nature of disseminated lesions was known to pathologists in the early nineteenth century particularly as described by Carswell, Cruveilhier and later, Frerichs, but J.M Charcot at the Salpêtrière later in the century is justly credited with the first serious study of the clinical and pathologic aspects of the disease. He collected 34 cases and set a foundation for understanding the disease. With neurosyphilis, multiple sclerosis formed much of the basis of early clinicopathologic correlation and the clinical method in neurology. It is therefore among the most venerable of neurologic diseases and one of the most important by virtue of its frequency, chronicity, and tendency to affect young adults. Cruveilhier (circa 1835), in his original description of the disease, attributed it to suppression of sweat, and since that time there has been endless speculation about the etiology. While many of the early theories are anachronistic in the light of present-day concepts, others are still of interest. There is little point in enumerating them here. The historical aspects can be found in the corresponding chapter of the text by Compston and colleagues.
Multiple sclerosis is a chronic condition characterized clinically by episodes of focal disorders of the optic nerves, spinal cord, and brain, which remit to a varying extent and recur over a period of many years and are usually progressive. The neurologic manifestations are protean, being determined by the varied location and extent of the demyelinating foci. Nevertheless, the lesions have a predilection for certain parts of the CNS, resulting in complexes of symptoms and signs and imaging appearances that can often be recognized as distinctive of MS as discussed in detail further on.
Typical features include weakness, paraparesis, paresthesias, loss of sight, diplopia, nystagmus, dysarthria, tremor, ataxia, impairment of deep sensation, and bladder dysfunction. The diagnosis may be uncertain at the onset and in the early years of the disease, when symptoms and signs point to a lesion in only one locus of the nervous system. Later, as the disease recurs and disseminates throughout the central nervous system, the diagnosis becomes quite certain. There may be a long period of latency (1 to 10 years or longer) between a minor initial symptom, which may not even come to medical attention, and the subsequent development of more characteristic symptoms. In most cases, there is initially a relapsing-remitting pattern, i.e., the signs and symptoms improve partially or completely, followed after a variable interval by the recurrence of the same abnormalities or the appearance of new ones in other parts of the nervous system. However, in fewer than half of patients, the disease takes the form a steadily progressive course, especially in patients older than 40 years of age at the time of onset (primary progressive MS). Or, as happens more often, an initially relapsing profile later becomes steadily progressive (secondary progressive MS).
A rule that had in the past guided clinicians is that the diagnosis of MS was not secure unless there was a history of remission and relapse and evidence on examination of more than one discrete lesion of the CNS. The advent of MRI and its capacity to identify clinically inevident lesions has replaced the exclusive dependence on clinical criteria for the diagnosis.
Before being sectioned, the brain and spinal cord generally show no evidence of disease, but the surface of the spinal cord may appear and feel uneven. Sectioning of the brain and cord discloses numerous scattered patches where the tissue is slightly depressed below the cut surface and stands out from the surrounding white matter by virtue of its pink-gray color (a result of loss of myelin). The lesions may vary in diameter from less than a millimeter to several centimeters; they principally affect the white matter of the brain and spinal cord, and do not extend beyond the root entry zones of the cranial and spinal nerves. It is because of their sharp delineation that they were called plaques by French pathologists.
The topography of the lesions is noteworthy. A periventricular localization is characteristic, but only where subependymal veins line the ventricles (mainly adjacent to the bodies and atria of the lateral ventricles). Other favored structures are the optic nerves and chiasm (but rarely the optic tracts) and the spinal cord, where pial veins lie next to or within the white matter. The lesions are distributed randomly throughout the brainstem, spinal cord, and cerebellar peduncles without reference to particular systems of fibers, but always confined predominantly to the white matter. In the cerebral cortex and central nuclear and spinal structures, the acute lesions destroy myelin sheaths but leave the nerve cells mostly intact. Severe and more chronic lesions, however, may destroy axons and neurons in the affected region, but the dominant lesion is still demyelinating.
The histologic appearance of the lesion depends on its age. Relatively recent lesions show a partial or complete destruction and loss of myelin throughout a zone formed by the confluence of many small, predominantly perivenous foci; the axons in the same region are relatively spared or less affected. There is a variable but usually slight degeneration of oligodendroglia, a variable astrocytic reaction, and perivascular and para-adventitial infiltration with mononuclear cells and lymphocytes as discussed in detail further on. Later, large numbers of microglial phagocytes (macrophages) infiltrate the lesions and astrocytes in and around the lesions increase in number and size. Long-standing lesions, on the other hand, are composed of thickly matted, relatively acellular glial tissue, with only occasional perivascular lymphocytes and macrophages; in such lesions, a few intact axons may still be found. In old lesions with interruption of axons, there may be descending and ascending wallerian degeneration of long fiber tracts in the spinal cord. Partial remyelination is believed to take place on undamaged axons and to account for incompletely demyelinated “shadow patches” (Prineas and Connell). A few of the most severe older lesions will have undergone cavitation, indicating that the disease process has affected not only myelin and axons but also supporting tissues and blood vessels. All gradations of histopathologic change between these two extremes may be found in lesions of diverse size, shape, and age, consistent with the extended clinical course.
The relatively ineffective remyelination of the MS plaque leaves in its wake denuded axons that are thinly myelinated, creating the just mentioned shadow plaques. Histologic evidence suggests that some of the oligodendrocytes are destroyed in areas of active demyelination but also that the remaining ones have little ability to proliferate. Instead, there is an influx of oligodendroglial precursor cells, which mature into oligodendrocytes and provide the remaining axons with new myelin. Probably the astrocytic hyperplasia in regions of damage and the persistent inflammatory response account for some of the inadequacy of the reparative process (see Prineas et al).
An insight into the complexity of the immunopathologic process can be appreciated in the analyses by Lucchinetti and colleagues (2000) of autopsy and brain biopsy specimens from patients with MS. They separated the lesions into four histologic subgroups: inflammatory lesions made up of T cells and macrophages alone (pattern I); an autoantibody lesion mediated by immunoglobulin and complement (pattern II); those characterized by apoptosis of oligodendrocytes and absence of immunoglobulin, complement, and with partial remyelination (pattern III); and those showing only oligodendrocyte dystrophy and no remyelination (pattern IV). Two features are of interest here. First, each case demonstrated only one pattern of pathology, suggesting that perhaps different pathophysiologic processes operated in each patient. Moreover, the last two histopathologic types were considered to represent a primary oligodendroglial cell degeneration. Some confirmation of a primary process in oligodendrocytes is the material from newly symptomatic lesions reported by Barnett and Prineas, in which there was loss of these cells. In addition, early lesions have been found to contain areas of demyelination within the cerebral cortex and these are often in contiguity with meningeal inflammatory infiltrates, or lymphoid follicles (Lucchinetti et al 2011, Howell et al).
The overall implication is that the pathologic characteristics of the chronic progressive type of MS may differ from those of the typical relapsing type (see further on). Most data suggest that antibody and complement-mediated myelin phagocytosis are the dominant mechanism of demyelination in MS. At the moment, we continue to conceptualize MS as mainly an inflammatory-immune process that targets central myelin along the lines of the observations of Adams and Kubik in their earlier studies, who were aware of the axonal and cortical changes in pathologic material they collected in the 1940s.
The incidence of MS is two or three times higher in women than in men but the basis of this fact is unclear, the best current explanation being that women are generally more susceptible to immune and inflammatory conditions. The incidence in children is very low; only 0.3 to 0.4 percent of all cases appear during the first decade. In an analysis of a small number of childhood-onset cases, Hauser and colleagues (1982) found no phenotypic differences between childhood and adult cases, but Renoux and colleagues analyzed a cohort of 394 patients who had MS with an onset at 16 years or younger and found that these patients took longer to reach states of irreversible disability, but did so at a younger age than patients with adult-onset MS. Beyond childhood, the risk of first developing symptoms of the disease rises steeply with age, reaching a peak at about 30 years, remaining high in the fourth decade, then falling off sharply and becoming low in the sixth decade. On this basis it has been pointed out that MS has a unimodal age-specific onset curve, similar to that of infectious and connective tissue diseases.
In a smaller number, the disease appears to develop in late adult life (late fifties and sixties). In such patients, early symptoms may have been forgotten or may never have declared themselves clinically (we have several times found the typical lesions of MS in aged autopsied individuals who had no history of neurologic illness). Gilbert and Sadler report five such cases and from their pathologic findings suggest that the true incidence of MS may be three times higher than the stated figures.
Although the cause of MS remains undetermined, a number of epidemiologic facts have been established and will eventually have to be incorporated in any hypothesis. The disease has a prevalence of less than 1 per 100,000 in equatorial areas; 6 to 14 per 100,000 in the southern United States and southern Europe; and 30 to 80 per 100,000 in Canada, northern Europe, and the northern United States. Mayr and colleagues reported an incidence of 8 and a prevalence of 177 cases per 100,000 in Olmstead County, Minnesota; this prevalence has been stable for approximately 30 years. A less-well-defined gradient exists in the southern hemisphere. Kurland’s studies indicated that there is a threefold increase in prevalence and a fivefold gradient in mortality rate between New Orleans (30 degrees north latitude) and Boston (42 degrees north) and Winnipeg (50 degrees north). In Japan, there is a similar although less distinct latitudinal gradient (the prevalence of MS there is much lower than in corresponding latitudes of North America and northern Europe).
The increasing risk of developing MS with higher and lower latitude has been confirmed by many epidemiologists following the work of Kurtzke (1975). In the United States, African Americans are at lower risk than whites at all latitudes, but both races show the same south-to-north gradient in risk, findings that invoked an environmental factor regardless of genetic predisposition. Supporting this view are the descriptions, by Kurtzke and Hyllested, of an “epidemic” of MS in the Faroe Islands of the North Atlantic. They found a much-higher-than-expected incidence of the disease, occurring as three separate outbreaks of decreasing extent between the years 1943 and 1973. (It should be pointed out that the largest outbreak consisted of only 21 cases.) It was their contention, confirmed by Poskanzer and colleagues, that the disease was the result of an unidentified infection introduced by British troops who occupied the islands in large numbers in the years immediately preceding the outbreak. Kurtzke and colleagues (1982) described a similar postwar epidemic in Iceland. The cause of these geographic distributions has been reinterpreted in terms of migration and population genetics rather than a number of other imputed causes, but they remain interesting (see Compston and Confavreaux for a complete discussion).
The role of Vitamin D and of sun exposure has become an area of related epidemiologic research. Some data suggest that the risk of MS is in part a result of a lack of exposure to these two related environmental features (Munger et al and van der Mei et al). Whether this partly explains the latitudinally graded risk is unclear. An observed seasonal fluctuation in the activity of established MS lesions may have a similar basis.
Several studies indicate that persons who migrate from a high-risk to a low-risk zone carry with them at least part of the risk of their country of origin and genetic makeup, even though the disease may not become apparent until 20 years after migration. Such a pattern has been demonstrated in both South Africa and Israel. Dean determined that the prevalence of MS in native-born white South Africans was 3 to 11 per 100,000, whereas the rate in immigrants from northern Europe was approximately 50 per 100,000, only slightly less than among the nonimmigrating natives of those countries. The data of Dean and Kurtzke indicate further that in persons who had immigrated before the age of 15, the risk was similar to that of native-born South Africans; whereas in persons who had immigrated after that age, the risk was similar to that of their birthplace. Alter and colleagues found that in the descendants of European immigrants born in Israel, the risk of MS was low, similar to that of other native-born Israelis, whereas among recent immigrants the incidence in each national group approached that of the land of birth. Again, the critical age of immigration appeared to be about 15 years. These older epidemiologic studies and others have suggested that MS is associated with particular localities rather than with a particular ethnic group in those localities, and implicate environmental factors but not to the exclusion of genetic susceptibility. However, more current studies suggest the opposite; that genetic factors in a population predominate.
A familial aggregation of MS is now well established. Approximately 15 percent of MS patients have an affected relative, with the highest risk of concurrence being observed in the patient’s siblings (Ebers, 1983). In a large population-based study carried out in British Columbia by Sadovnick and colleagues (1988), it was found that almost 20 percent of index cases had an affected relative, again with the highest risk in siblings. In a subsequent study, Sadovnick and colleagues (1996) sought to determine the degree of heritability of MS by comparing the risk of disease in the half-sibs (one biologic parent in common) of affected individuals with the risk in full sibs; the risk for full sibs was two to three times greater than for half-sibs and they interpreted these results as clearly genetic in basis.
The case for heritability is further supported by studies of twins in whom one of each pair is known to have MS. In the most extensive of these studies (Ebers et al), the diagnosis was verified in 12 of 35 pairs of monozygotic twins (34 percent) and in only 2 of 49 pairs of dizygotic twins (4 percent). Furthermore, in two additional sets of monozygotic twins who were clinically normal, lesions were detected by MRI. The concordance rate in dizygotic pairs is similar to that in nontwin siblings. Despite these provocative findings, no consistent pattern of mendelian inheritance has emerged. Of course, one must not assume that all diseases with an increased familial incidence are hereditary in that instances of the same condition in several members of a family may simply reflect an exposure to a common environmental agent. Paralytic poliomyelitis, for example, was about eight times more common in immediate family members than in the population at large.
Further evidence of a genetic factor in the causation of MS is the finding that certain histocompatibility locus antigens (HLAs) are more frequent in patients with MS than in control subjects. The strongest association is with the DR locus on chromosome 6. Other HLA haplotypes that are overrepresented in MS (HLA-DR2 and, to a lesser extent, -DR3, -B7, and -A3) are thought to be markers for an MS “susceptibility gene”—possibly an immune response gene. The presence of one of these markers increases the risk that an individual will develop MS by a factor of 3 to 5. These antigens may indeed prove to be related to the frequency of the disease, but their presence is not invariable and their exact role is far from clear. A genome-wide association study identified several alleles, interleukin (IL)-2Rα, and IL7Rα in addition to the previously established HLA loci, as heritable risk factors for MS (International Multiple Sclerosis Genetics Consortium). These findings, although they apply to a small number of individuals, support the concept that dysregulation of the immune response is a factor in the risk for developing MS.
The low conjugal incidence of MS, on the other hand, indicates that any common exposure to an inciting infection or environmental agent must occur early in life. To test this hypothesis, Schapira and coworkers determined the periods of common exposure (common habitation periods) in members of families with two or more cases. From this they calculated the mean common exposure to have happened before 14 years of age, with a latency of about 21 years—figures that are in general agreement with those derived from the migration studies quoted above.
Several studies from northern Europe and Canada suggest that the likelihood of developing MS is somewhat greater among rural than among urban dwellers; studies of American army personnel indicate the opposite (Beebe et al). A number of surveys in Great Britain intimate that the disease is more frequent in the higher socioeconomic groups than in the lower ones. Yet in the United States, no clear relationship has been established to the poverty or social deprivations that are part of a low socioeconomic status. Numerous other environmental factors (surgical operations, trauma, anesthesia, exposure to household pets [small dogs], cobalamin deficiency or resistance, mercury in silver amalgam fillings in teeth), and Lyme disease have been proposed but are unsupported by firm evidence and probably are mostly spurious associations.
These epidemiologic data point to both a genetic susceptibility and some environmental factor that is encountered in childhood that, after years of latency, evokes the disease. Over the years, data favoring an infection, most often viral as the triggering factor, have had periods of support (see above). A body of indirect evidence has been marshaled in support of this idea, based largely on alterations in humoral and cell-mediated immunity to viral agents. To this day, however, no virus (including all known members of the human retrovirus family) has been seen in, or isolated from, the tissues of patients with MS despite innumerable attempts to do so. Moreover, no satisfactory viral model of MS has been produced experimentally. The bacterial agents Chlamydia pneumoniae and Borrelia burgdorferi (the agent of Lyme disease) and herpesvirus type 6 have been similarly implicated by the finding of their genomic material in MS plaques, but the evidence for their direct participation in the disease is, at the moment, not compelling.
If, indeed, some obscure infection is the initial event in the genesis of MS, then a secondary factor must be operative in later life to reactivate the disease and cause exacerbations. One view is that this secondary mechanism is an autoimmune reaction attacking some component of myelin and, in its most intense form, destroying all tissue elements, including axons. Several lines of argument have been advanced in support of this view. One is inclined to draw an analogy between the lesions of MS and those of acute disseminated encephalomyelitis, which is almost certainly an autoimmune disease of delayed hypersensitivity type (see further on). Also in support of this possibility is the finding of antibodies to specific myelin proteins—for example, myelin basic protein (MBP)—in both the serum and cerebrospinal fluid (CSF) of MS patients, and these antibodies, along with T cells that are reactive to MBP and to other myelin proteolipids, increase with disease activity; moreover, MBP cross-reacts to some extent with measles virus antibodies. The arguments that a chronic viral infection reactivates and perpetuates the disease are, however, less convincing than those proposing a role for viruses in the initiation of the process in susceptible individuals.
The relative roles of humoral and cellular factors in the production of MS plaques are not fully understood. The deposition of immunoglobulin in the plaques of patients with acute and relapsing–remitting disease, but not in the plaques of those with progressive MS, was alluded to earlier. That the humoral immune system is involved is evident from the presence in the CSF of most patients of oligoclonal immune protein antibodies, which are produced by B lymphocytes within the CNS. Sera from patients with MS (and some normal controls), when added to cultures of nervous system tissue from newborn mice in the presence of complement, can damage myelin, inhibit remyelination, and block axonal conduction. Antibodies to oligodendrocytes are present in the serum of up to 90 percent of patients in some studies, but far less frequently in others.
Autoantibodies have been found inconsistently that are directed against myelin oligodendrocyte glycoprotein (MOG) and MBP. It has also been demonstrated that subsets of T cells (CD41 Th2 cells) are activated by MBP and MOG to activate B cells, the production of oligoclonal bands and membrane attack complexes, and the release of cytokines (tumor necrosis factor-alpha [TNF-α], interleukins, interferon-gamma [IFN-γ ]). The inflammatory process erodes the blood–brain barrier and ultimately destroys both oligodendroglia and axons. The eventual functional outcome reflects both the activity of this inflammatory cascade and the degree of axonal damage. In other cases, there may be a compromise of oligodendroglial function and axonal degeneration in the absence of prominent inflammation. Many times, one or another putative antigenic target has been found by immunologic techniques in one laboratory, only to fail to be replicated by another group. None of these provide a unifying etiology for the disease but the humoral aspects may provide insights particularly into the pauci-inflammatory type of oligodendrocyte degeneration that characterizes some lesions, as discussed in the section on pathology.
Nevertheless, most immunologists currently subscribe to the notion that MS is mediated by a T-cell sensitization to some component of myelin. This idea is supported by numerous lines of evidence, including the observation that T cells initiate the lesions of experimental allergic encephalomyelitis (EAE), which is assumed to be an approximate animal model of MS, as suggested originally by Waksman and Adams. It has been difficult, however, to produce a relapsing experimental form of the illness that would simulate MS. Although the entry of autoreactive T cells into the CNS results in a perivascular inflammatory reaction, its relationship to MS is unclear. Conceivably, intense T-cell stimulation is in itself sufficient to induce demyelination but it is also possible that the primary target of the immune reaction is the myelin sheath or some component thereof and that the T-cell infiltration is a reaction to demyelination. Most investigators believe that an additional insult is required, as illustrated by the EAE animal model, in which myelin alone is not a sufficient factor but always requires an adjuvant immune stimulus. EAE is clearly an imperfect model; it is not a naturally occurring disease but one in which a demyelination of the CNS is induced in susceptible animals in a single episode by autologous myelin antigens. The inducing antigen in EAE is known, whereas the putative antigens in MS are not.
Also incorporated into most theories of the immune pathogenesis is an alteration of the blood–brain barrier, represented by adhesion of lymphocytes to endothelial cells in the nervous system. Whether this is an active interaction or a passive event triggered by antigenic attraction is not clear; nonetheless, these cell–vascular interactions have been incorporated into pathogenic theories and are the basis of newer treatments for MS. Always in the background is the element of genetic susceptibility, presumably making certain individuals prone to these immunologic events as noted in the earlier sections.
The foregoing data notwithstanding, the immune mechanisms in MS are not fully specified and the autoimmune hypothesis is not beyond challenge. It is noteworthy that the prevalence of other diseases of presumed autoimmune origin in some series is no higher in MS patients than in the general population (De Keyser). However, various epidemiologic studies differ on this point and some have found an increase in autoimmune diseases in affected patients and in their families.
The main physiologic effect of demyelination is to impede saltatory electrical conduction of nerve impulses from one node of Ranvier, where sodium channels are concentrated, to the next node. The resulting failure of electrical transmission is thought to underlie most of the abnormalities of function resulting from demyelinating diseases of both the central and peripheral nerves. As an example, the delay in electrical conduction in the optic nerve (found by using pattern-shifting visual stimuli in MS patients) raises a number of points about the pathophysiology of demyelination. When the demyelinating process is acute and reversible within a few days, the block in nerve fiber conduction is obviously physiologic rather than pathologic; in such a brief period, recovery is unlikely to have been a result of remyelination; recovery is probably a result of subsidence of the edema and acute inflammatory changes in and around the lesion. Remyelination probably does occur, but it is a slower process and partial at best, and its functional effects in the CNS are possibly expressed as a slowing of nerve conduction, which, if present in an eye with normal vision, may account for the reduction in flicker fusion and in the perception of multiple visual stimuli (Halliday and McDonald). It also explains one of the typical symptoms of optic neuritis—reduction in the intensity (desaturation) of the color red. It is clear, however, that many of the plaques in the cerebral hemispheres that are visualized on MRI are unaccompanied by corresponding symptoms. Either there has been complete remyelination in these plaques, sufficient to support clinical functioning, or, in the acute stage, the plaque may represent edema rather than demyelination.
Another typical feature of MS is the temporary induction, by heat or exercise, of symptoms such as unilateral visual blurring (the Uhthoff phenomenon) or tingling and weakness of a limb (the basis of the hot-tub test used in previous years). This has been shown experimentally to represent an extreme sensitivity of conduction in demyelinated nerve fibers to an elevation in temperature. A rise of only 0.5°C (0.9°F) can block electrical transmission in thinly myelinated or demyelinated fibers. Likewise, hyperventilation slows conduction of the visual evoked response, an effect that is rarely perceived by the patient. The remarkable sensitivity of demyelinated and remyelinated regions to subtle metabolic and environmental changes provides an explanation for the rapid onset of symptoms in some patients and the apparent fluctuations of MS that show no laboratory evidence of active inflammatory changes in the CNS. Smoking, fatigue, hyperventilation, and a rise in environmental temperature are all capable of briefly worsening neurologic functioning and are easily confused with relapses of disease.
Weakness or numbness, sometimes both, in one or more limbs is the initial symptom in about half the patients. Symptoms of tingling of the extremities and tight band-like sensations around the trunk or limbs are commonly associated and are probably the result of involvement of the posterior columns of the spinal cord. The symptoms generally appear over hours or days, at times being so trifling that they are ignored, and less often, coming on so acutely and prominently as to bring the patient urgently to the doctor. The resulting clinical syndromes vary from a mere dragging or poor control of one or both legs to a spastic or ataxic paraparesis. The tendon reflexes are retained and later become hyperactive with extensor plantar reflexes; varying degrees of deep and superficial sensory loss may be associated. It is a useful adage that the patient with MS presents with symptoms in one leg but with signs in both; the patient will complain of weakness, incoordination, or numbness and tingling in one lower limb and prove to have bilateral Babinski signs and other evidence of bilateral corticospinal and posterior column disease.
There are, in addition, several syndromes that are typical of multiple sclerosis and may be the initial manifestations. These common modes of onset are: (1) optic neuritis, (2) transverse myelitis, (3) cerebellar ataxia, and (4) brainstem syndromes (vertigo, facial pain or numbness, dysarthria, diplopia). When these are unaccompanied by other features of MS, they are termed “clinically isolated syndrome” (CIS) but they are often aspects of the established disease as well. In the initial phases of the illness, they may pose diagnostic questions, as they also certainly occur with numerous diseases other than MS.
Flexion of the neck may induce a tingling, electric-like feeling down the shoulders and back and, less commonly, down the anterior thighs. This phenomenon is known as the Lhermitte sign, although it is more a symptom than a sign and was originally described by Babinski in a case of cervical cord trauma. Lhermitte’s contribution was to draw attention to the frequent occurrence of this phenomenon in MS. It is probably attributable to an increased sensitivity of demyelinated axons to the stretch or pressure on the spinal cord induced by neck flexion, but it occurs in other conditions such as cervical spondylosis.
McAlpine and coworkers (1972) analyzed the mode of onset in 219 patients and found that in 20 percent the neurologic symptoms were fully developed in a matter of minutes, and, in a similar number, in a matter of hours. In approximately 30 percent the symptoms evolved more slowly, over a period of a day or several days, and in another 20 percent more slowly still, over several weeks to months. In the remaining 10 percent the symptoms had an insidious onset and slow, steady, or intermittent progression over months and years. The typical relapsing–remitting pattern of disease is more likely to appear in patients who are younger than 40 years of age. Certain paroxysmal symptoms and signs may occur in the established phase of the disease and discussed further on. The inflammatory process of MS affects no organ system other than the CNS.
In approximately 25 percent of all MS patients (and possibly in a larger proportion of children), the initial manifestation is an episode of optic neuritis. It will be recalled that the optic nerve is in fact a tract of the brain, and involvement of the optic nerves is therefore consistent with the rule that lesions of MS are confined to the CNS. Characteristically, over a period of several days, there is partial or total loss of vision in one eye. Many patients, for a day or two before the visual loss, experience pain within the orbit, worsened by eye movement or palpation of the globe. Rarely, the visual loss is steadily progressive for several weeks, mimicking a compressive lesion or intrinsic tumor of the optic nerve (Ormerod and McDonald). Usually a scotoma involving the macular area and blind spot (cecocentral) can be demonstrated, but a wide variety of other field defects may occur, rarely even hemianopic involvement (sometimes homonymous). In some patients, both optic nerves are involved, either simultaneously or, more commonly, within a few days or weeks of one another, and at least one in eight patients will have repeated attacks.
Serial examinations may disclose evidence of swelling or edema of the optic nerve head (papillitis) in about a tenth of the patients. The occurrence of papillitis depends on the proximity of the demyelinating lesion to the nerve head. As emphasized in Chap. 13, papillitis can be distinguished from the papilledema of increased intracranial pressure by the severe and acute visual loss that accompanies only the former. More often, the optic nerve head appears normal or nearly so; this represents retrobulbar neuritis. Subtle manifestations of optic nerve affection, such as an afferent pupillary defect, atrophy of retinal nerve fibers, or sheathing of retinal veins and abnormalities of the visual evoked response (Chap. 2), should be sought in patients who have no visual complaints but are suspected of having MS. In cases of substantial visual loss, there is a diminished pupillary response to light (afferent pupillary paralysis) and instability of the direct pupillary response but the pupil is not dilated in ambient light. If the optic neuritis is unilateral, the consensual light reflex from the normal eye is retained. (Demyelination of the third nerve in its brainstem course, however, may be associated with a fixed enlargement of the pupil.) Visual evoked potentials and optical coherence tomography (OCT) may be useful in detecting optic neuritis, as discussed in a later section and in Chap. 2.
As noted in Chap. 13, about half of patients with optic neuritis recover completely, and most of the remaining ones improve significantly, even those who present initially with profound visual loss and, later, pallor of the optic disc (Slamovitis et al). Any pain in the globe is short-lived and persistent pain should prompt an evaluation for local disease. In a cohort of 397 patients enrolled in the Optic Neuritis Treatment Trial and examined 5 years after the initial attack of optic neuritis, visual acuity had returned to 20/25 or better in 87 percent of patients and to 20/40 or better in 94 percent—even if there had been a recurrence of optic neuritis during the 5-year period. Moreover, the mode of treatment did not appear to influence the outcome. Dyschromatopsia, generally taking the form of a perceived desaturation of colors, frequently persists as does the Pulfrich effect, wherein an object such as a pendulum that is swinging perpendicular to the patient’s line of sight, appears to moving in a three-dimensional, circular motion.
When improvement occurs, it usually begins within 2 weeks of onset, as is true of most acute manifestations of MS, perhaps sooner with corticosteroid treatment. Once improvement in neurologic function begins, it may continue for several months.
More than one-half of adult patients who present with optic neuritis will eventually develop other signs of MS. The prospective investigation of Rizzo and Lessell showed that MS developed in 74 percent of women and 34 percent of men by the fifteenth year after onset of visual loss; similar results were reported by the Optic Neuritis Study Group (Beck et al, 2003). The risk is much lower if the initial attack of optic neuritis occurs in childhood (26 percent developed after 40 years of followup [Lucchinetti et al 1997]); this suggests that some instances of the childhood disease may be of a different type, perhaps viral or postinfectious. The longer the period of observation and the greater the care given to detection of mild cases, the greater the proportion of patients who are found to develop signs of MS; however, most do so within 5 years of the original attack (Ebers, 1985; Hely et al). In fact, in many patients with clinically isolated optic neuritis, MRI has disclosed lesions of the cerebral white matter—suggesting that dissemination, albeit asymptomatic, had already occurred and thereby establishing the diagnosis of MS (Jacobs et al, 1986; Ormerod et al). The Optic Neuritis Study Group has made the point, well known to neurologists, that the recurrence of optic neuritis greatly increases the chances of developing MS. Of practical value is the observation, in the study by Beck and colleagues (2003), that the risk of relapsing-remitting MS is also considerably lower (22 percent at 10 years) if the cranial MRI fails to reveal demyelinating lesions.
It is unclear whether optic neuritis that occurs alone and is not followed by other evidence of demyelinating disease is simply a restricted form of MS or a manifestation of some other disease process, such as postinfectious encephalomyelitis. By far the most common pathologic basis for optic neuropathy is demyelinating disease, although it is known that a vascular lesion or compression of an optic nerve by a tumor or mucocele may cause a central or cecocentral scotoma that is indistinguishable from the defect of optic neuritis. Also, there may be a special form of chronic relapsing optic neuritis that is the result of an undefined granulomatous process such as sarcoid, as suggested by Kidd and colleagues. Uveitis and sheathing of the retinal veins are other ophthalmic disorders that occur with higher than expected incidence in patients with MS. The retinal vascular sheathing is caused by T-cell infiltration, identical to that in typical plaques, but this is an unusual finding, because the retina usually contains no myelinated fibers (Lightman et al). Optic neuritis is, of course, a common feature in neuromyelitis optica (Devic disease), discussed in a later section.
The treatment of optic neuritis is discussed further on.
This is the common designation for an acutely evolving inflammatory–demyelinating lesion of the spinal cord, which proves in many, but not all, instances to be an expression of MS. In this sense, the myelitic lesion is analogous to that of optic neuritis. The term transverse in relation to the myelitis is somewhat imprecise, implying that all of the elements in the cord are involved in the transverse plane, usually over a short vertical extent. Instead, in MS, the spinal cord signs are asymmetrical and incomplete and involve only a part of the long ascending and descending tracts, i.e., paraplegia and complete sensory loss are unusual.
Clinically, the illness is characterized by a rapidly evolving (several hours or days) symmetrical or asymmetrical paraparesis or paraplegia, ascending paresthesia, loss of deep sensibility in the feet, a sensory level on the trunk, sphincteric dysfunction, and bilateral Babinski signs. The CSF shows a modest number of lymphocytes and increase in total protein but both may be normal early in the illness. As many as one-third of patients report an infectious illness in the weeks preceding the onset of neurologic symptoms, in which case a monophasic postinfectious demyelinating disease rather than MS is the likely cause of the myelitis. The MRI usually shows indications of focal demyelination in the spinal cord at the appropriate level and there may be enhancement with gadolinium infusion, but neither of these findings is invariable. The lesions, as shown in Fig. 36-1 (lower right panel), are almost indistinguishable from those of postinfectious myelitis. In those instances associated with existing MS, even if not previously symptomatic, MRI of the cerebral hemispheres will show lesions consistent with demyelination; the absence of such lesions, however, does not ensure that the myelitic illness is monophasic and will not evolve to MS. Some cases progress to a necrotic myelopathy, with or without optic neuropathy, that is an expression of neuromyelitis optica, as discussed in a later section.
Figure 36-1.
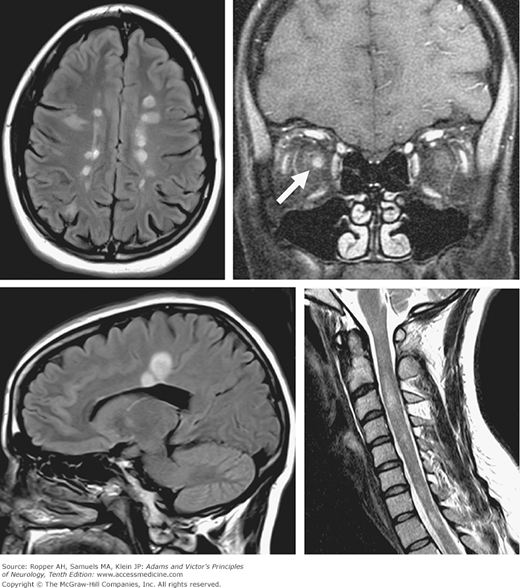
MRI in multiple sclerosis. Upper left, axial T2-FLAIR image showing multiple discrete periventricular hyperintense plaques, as well as two subcortical plaques in the right frontal and parietal lobes. Upper right, coronal T1-post gadolinium image showing abnormal enhancement of the right optic nerve in a case of acute optic neuritis (arrow). Lower left, sagittal T2-FLAIR image showing two hyperintense plaques emanating radially from the body of the corpus callosum (“Dawson fingers”). Lower right, sagittal T2 MRI showing multiple discrete hyperintense plaques within the cervical spinal cord. The lesion at C3 is acute with accompanying expansion of the cord. The lesion at the T1 level of the cord is chronic and shows cord atrophy.
Fewer than half the patients have evidence of an asymptomatic demyelinating lesion elsewhere in the nervous system or develop clinical evidence of dissemination within 5 years of the initial attack of acute myelitis (Ropper and Poskanzer). Not entirely in accord with our experience is the analysis of subgroups in a trial of interferon therapy conducted by Beck and colleagues (2002), in which the cumulative probability of developing MS after 2 years was similar after either optic neuritis or transverse myelitis. Our sense has been that acute transverse myelitis is somewhat less often an initial expression of MS than is optic neuritis.
A special problem is presented by patients with recurrent myelitis at one level of the spinal cord but in whom no other signs of demyelinating disease can be found by careful clinical examination or MRI. Some of them may even have oligoclonal bands in the CSF, which are commonly associated with MS (see further on). Enough cases of this limited nature have come to our attention to permit the conclusion that there is a recurrent form of spinal cord MS in which cerebral dissemination is infrequent (Tippett et al). Isolated recurrent myelitis or myelopathy occurs also with lupus erythematosus, sarcoidosis, Sjögren syndrome, mixed connective tissue disease, and the antiphospholipid antibody syndrome or in the presence of other autoantibodies, as well as with dural and cord vascular fistulas and arteriovenous malformations. An analogous situation pertains in respect to some instances of optic neuritis—repeated attacks that remain confined to the optic nerve.
Other aspects of transverse myelitis are discussed in Chap. 44, and later in this chapter.
Like the modes of onset cited above, other early manifestations of MS are unsteadiness in walking, brainstem symptoms (diplopia, vertigo, vomiting), paresthesias or numbness of an entire arm or leg, facial pain often simulating tic douloureux, and disorders of micturition. Vertigo of central type is also a frequent initial sign of MS, but it more often appears in established cases. Discrete manifestations such as hemiplegia, pain syndromes, facial paralysis, deafness, or seizures occur in an only small proportion of cases. Most often the disease presents with more than one of the aforementioned symptoms almost simultaneously or in rapid succession. Another relatively isolated syndrome, occurring mainly in older women, is a slowly progressive cervical myelopathy with weakness and ataxia. This is particularly difficult to differentiate from cervical spondylosis.
Not infrequently a prominent feature of the disease is nystagmus and ataxia, with or without weakness and spasticity of the limbs, a syndrome that reflects involvement of the cerebellar and corticospinal tracts. Ataxia of cerebellar type can be recognized by scanning speech, rhythmic instability of the head and trunk, intention tremor of the arms and legs, and incoordination of voluntary movements and gait, as described in Chap. 5. The combination of nystagmus, scanning speech, and intention tremor is known as the Charcot triad. While this group of symptoms is often seen in the advanced stages of the disease, most neurologists would agree that it is not a common mode of presentation. The most severe forms of cerebellar ataxia, in which the slightest attempt to move the trunk or limbs precipitate a violent and uncontrollable ataxic tremor, are observed among patients with long-standing MS. The responsible lesion probably lies in the tegmentum of the midbrain and involves the dentatorubrothalamic tracts and adjacent structures. Cerebellar ataxia may be combined with sensory ataxia, owing to involvement of the posterior columns of the spinal cord or medial lemnisci of the brainstem. In most cases of this type, the signs of spinal cord involvement ultimately predominate; in others, the cerebellar signs are more prominent.
Diplopia is another common presenting complaint. It is most often a result of involvement of the medial longitudinal fasciculi, producing an internuclear ophthalmoplegia (see Chap. 14). The signs are characterized by paresis of the medial rectus on attempted lateral gaze, with a coarse nystagmus in the abducting eye; in MS, this abnormality is usually bilateral (unlike small pontine infarcts, which cause a unilateral internuclear ophthalmoplegia [INO]). As a corollary, the presence of bilateral internuclear ophthalmoplegia in a young adult is virtually diagnostic of MS. Occasionally, internuclear ophthalmoplegia in one direction is combined with a horizontal gaze paresis in the other, although this “one-and-a-half syndrome” is more typical of brainstem stroke. Other palsies of gaze (a result of interruption of supranuclear connections) or palsies of individual ocular muscles (because of involvement of the ocular motor nerves in their intramedullary course) also occur, but less frequently. Additional manifestations of brainstem involvement include myokymia or paralysis of facial muscles, deafness, tinnitus, vertigo—as noted above, vomiting (vestibular connections), and, rarely, stupor and coma. The occurrence of transient facial hypesthesia or anesthesia or of trigeminal neuralgia in a young adult should always suggest the diagnosis of MS implicating the intramedullary fibers of the fifth cranial nerve.
Dull, aching, but otherwise nondescript pain in the low back is a common complaint, but its relation to the lesions of MS is uncertain. Infrequently, there is sharp, burning, poorly localized, or lancinating radicular pain, localized to a limb or discrete part of the trunk. Nevertheless, these types of pains, presumably caused by demyelinating foci involving the dorsal root entry zones, have a few times been the presenting feature of the disease or have appeared at a later time in established cases (see Ramirez-Lassepas et al for a discussion of pain in MS).
When the diagnosis of MS has become virtually certain, a number of clinical syndromes are observed to occur with regularity. Approximately one-half of the patients will manifest a clinical picture of mixed or generalized type with signs pointing to involvement of the optic nerves, brainstem, cerebellum, and spinal cord—specifically signs relating to the posterior columns and corticospinal tracts. Another 30 to 40 percent will exhibit only varying degrees of spastic ataxia and deep sensory changes in the extremities, i.e., essentially a spinal form of the disease. In either case, an asymmetrical spastic paraparesis with some degree of impaired joint position and vibration sense in the legs is probably the most common manifestation of progressive MS. A predominantly cerebellar or brainstem–cerebellar form occurs in approximately 5 percent of cases. Thus the mixed and spinal forms together have made up at least 80 percent of our clinical material.
It has become evident that some degree of cognitive impairment, and probably a progressive decline, is present in perhaps one-half of patients with long-standing MS. The process is characterized by reduced attention, diminished processing speed and executive skills, and memory decline, while language skills and other intellectual functions are preserved, features that have been subsumed under “subcortical dementia,” as discussed in Chap. 21. Other mental disturbances, such as a loss of retentive memory, a global dementia, or a confusional–psychotic state, also occur in limited cases in the advanced stages of the disease, but we have found this degree of deterioration to be exceptional. The decline in cognitive functions correlates with quantifiable MRI measurements, particularly loss of white matter volume, thinning of the corpus callosum, and brain atrophy (reviewed by Bobholz and Rao).
Traditional teaching has probably overemphasized the frequency of euphoria, a pathologic cheerfulness or elation that seems inappropriate in the face of the obvious neurologic deficit. (Charcot spoke of this phenomenon as “stupid indifference” and Vulpian as “morbid optimism.” It has often been referred to as “la belle indifférence.”) Some patients do show this abnormality, usually in association with other signs of cerebral impairment. In some instances, it is manifestly a part of the syndrome of pseudobulbar palsy. A much larger number of patients, however, are depressed, irritable, and short-tempered, sometimes as a reaction to the disabling features of the disease but also apparently as a primary effect of the brain disease; the incidence of depression has been estimated to be as high as 25 to 40 percent in some series. Dalos and coworkers, in comparing MS patients with a group of traumatic paraplegics, found a significantly higher incidence of emotional disturbance in the former group, especially during periods of relapse. As mentioned above, the cognitive impairment is in keeping with what has been ascribed to “subcortical dementia” (see Chap. 21) but demyelination in the cortical layers is increasingly being recognized as a possible basis for dementia in MS. Loss of the volume of gray matter, for example, appears to be predictive of dementia as much as loss of central white matter. Either can give rise to global cerebral atrophy.
Symptoms of bladder dysfunction, including hesitancy, urgency, frequency, and incontinence, occur commonly with spinal cord involvement. Urinary retention, as a result of damage to sacral segments of the cord is less frequent (see Fig. 26-4). These symptoms are often associated with erectile dysfunction, a symptom that the patient may not report unless specifically questioned in this regard.
Paroxysmal attacks of neurologic deficit, lasting a few seconds or minutes and sometimes recurring many times daily, are relatively infrequent but well-recognized features of MS (see Mathews and also Osterman and Westerbey). Usually the attacks occur during the course of relapsing and remitting phase of the illness, rarely as an initial manifestation. These clinical phenomena are referable to any part of the CNS but tend to be stereotyped in an individual patient. The most common phenomena are dysarthria and ataxia, paroxysmal pain and dysesthesia in a limb, flashing lights, paroxysmal itching, or tonic “seizures”, taking the form of flexion (dystonic) spasm of the hand, wrist, and elbow with extension of the lower limb. The paroxysmal symptoms, particularly the tonic spasms, may be triggered by sensory stimuli or can be elicited by hyperventilation. On a few occasions we have seen dystonic hand and arm spasms as the first symptoms; an acute plaque was detected in the opposite internal capsule. In advanced cases, the spasms may involve all four limbs and even a degree of opisthotonos. The cause of paroxysmal phenomena is uncertain. They have been attributed by Halliday and McDonald to ephaptic transmission (“cross-talk”) between adjacent demyelinated axons within a lesion.
These transitory symptoms appear suddenly, may recur frequently for several days or weeks, sometimes longer, and then remit completely, i.e., they exhibit the temporal profile of a relapse or an exacerbation. It is sometimes difficult to determine whether they represent an exacerbation or a new lesion. Years ago, Thygessen pointed out, in an analysis of 105 exacerbations in 60 patients, that there were new symptoms in only 19 percent; in the remainder there was only a recurrence of old symptoms. Another problem is that the original lesion may have been asymptomatic. This is most obviously reflected in the many patients who are found to have impaired visual evoked responses but have never had symptomatic visual changes. Thus, new symptoms and signs may be manifestations of previously formed but asymptomatic plaques. However, the observations of Prineas and Connell indicate that symptoms and signs may progress without the appearance of new plaques. These and other factors need to be taken into consideration in evaluating the clinical course of the illness and the effects of a therapeutic program (see Poser, 1980). Carbamazepine is usually effective in controlling such spontaneous attacks, and acetazolamide blocks the painful tonic spasms that are elicited by hyperventilation.
Unusually severe fatigue is another peculiar symptom of MS; it is often transient and more likely to occur when there is fever or other evidence of disease activity but it can be a persistent complaint and a source of considerable distress. Depression may play a role in these recalcitrant cases, although the response to pharmacologic agents suggests that these two aspects of the disease are dissociable. Thus, antidepressants often do not improve fatigue, whereas drugs that alleviate fatigue, such as modafinil and amantadine, do not function as antidepressants.
A number of other interesting manifestations of MS have come to attention over the years and have given rise to difficulties in diagnosis. The occurrence of typical tic douloureux in young patients has already been mentioned; only their young age and the bilaterality of the pain in some of them raised the suspicion of MS, confirmed later by sensory loss in the face and other neurologic signs. It is notable, however, that facial palsy along the lines of Bell’s palsy is almost never a sign of MS. Brachial, thoracic, or lumbosacral pain consisting mainly of thermal and algesic dysesthesias was a source of puzzlement in several of our patients until additional lesions developed. Other types of pain in MS have been addressed earlier.
In two of our cases, the relatively acute occurrence of a right hemiplegia and aphasia first raised the probability of a cerebrovascular lesion; in still others, a more slowly evolving hemiplegia had led to an initial diagnosis of a cerebral glioma. The dystonic and paroxysmal symptoms are mentioned earlier; they do not typically bring the diagnosis of MS to mind.
There may be a slightly increased incidence of seizures in patients with MS but the frequency of the problem varies greatly among studies. It should be emphasized that seizures are usually in relation to an obvious cerebral lesion and advanced disease of many years duration. Seizures at an early stage of illness are almost always attributable to previous head injury, idiopathic epilepsy, or withdrawal of sleep medication, but not to MS.
Several times we have seen coma during relapse of longstanding MS, and in each instance it continued to death. In one case it occurred in a 64-year-old woman who had had two previous episodes of nondisabling spinal MS at 30 and 44 years of age. A confusional state with drowsiness was the initial syndrome in another patient whom we saw later with a relapse involving the cerebellum and spinal cord. Another unusual syndrome is one of slow intellectual decline with slight cerebellar ataxia. The chronic progressive form of MS is addressed below.
A variety of events occurring immediately before the initial symptoms or exacerbations of MS have been invoked as precipitating factors. The most common are infection, trauma, and pregnancy. However, in our view, none of these has been convincingly related to an increased risk of new attacks of MS, but there is little question that some febrile illnesses such as urinary infections can exaggerate the existing symptoms. The issue of truly precipitating a relapse as a result of a nondescript febrile illness is not resolved. Nonetheless, we have had experience with two patients who regularly had acute exacerbations of MS following each outbreak of labial genital herpes. The incidence of respiratory, urinary, or gastrointestinal viral infections that precede the onset or exacerbations of the disease varies greatly in different series, from 5 to 50 percent. The swine influenza vaccine, which was given to 45 million persons in the United States in late 1976, caused a slight increase in the incidence of Guillain-Barré disease but not of MS (Kurland et al), and more recent surveys of immunization programs, such as the one by Confavreux and colleagues (2001), have had similar results.
The possible role of trauma in precipitating MS is more difficult to assess. McAlpine and Compston found that the incidence of trauma within a 3-month period preceding the onset of MS was slightly greater than in a control group of hospital patients. Furthermore, there appeared to be a relationship between the site of the injury and the site of initial symptoms, particularly in patients who developed symptoms within a week of injury. We do not find this evidence convincing, particularly when given as an explanation for a large number of attacks. Other forms of trauma (including lumbar puncture and general surgical procedures) that occur after the onset of the neurologic disorder have not been shown to have an adverse effect on the course of the illness. Matthews, who has extensive personal experience with survivors of penetrating head wounds, did not find a single instance of MS among them. One of the most meaningful prospective studies of the relation of physical injury to MS is that of Sibley and colleagues, who followed 170 MS patients and 134 controls for an average of 5 years, during which they recorded all (1,407) instances of trauma and measured their effects on exacerbation rate and progression of the disease. With the possible exception of a case or two of electrical injury, there was no correlation between traumatic episodes and exacerbations. The current authoritative view on this subject is that the coincidence of trauma and new or exacerbated MS is incidental.
Certain other epidemiologic data have a bearing on this subject. There are, in the United States, 250,000 to 350,000 cases of physician-diagnosed MS (Anderson et al). Also, a study from the National Center for Health Statistics has determined that trauma sufficiently severe to be recalled at a periodic health examination occurs in one-third of the population of the United States (some 83 million persons) each year. Moreover, MS patients suffer physical injuries two or three times more often than normal persons (Sibley et al). In light of these data, it is perhaps not surprising that a traumatic event and an exacerbation should sometimes coincide, quite by chance.
Issues related to MS and pregnancy are addressed in a later section.
Several variants of MS present special problems that are addressed in this and in later section.
Rarely, MS takes a rapidly progressive and highly malignant form; Marburg’s name has been attached to this variant. A combination of cerebral, brainstem, and spinal manifestations evolves over a few weeks, rendering the patient stuporous, comatose, or decerebrate with prominent cranial nerve and corticospinal abnormalities. Death may end the illness within a few weeks to months without any remission having occurred, or there may be partial recovery, as noted below. At autopsy the lesions are of macroscopic dimensions, in essence very large acute plaques of MS. The only difference from the usual form of MS is that many plaques are of the same age and the confluence of many perivenous zones of demyelination is more obvious. Two of our most striking examples of this rapidly fatal form were in a 6-year-old girl and a 16-year-old boy, both of whom died within 5 weeks of the onset of symptoms. Another was a 30-year-old man who lived 2 months. In none of them had there been a preceding exanthem or inoculation or any symptoms suggestive of demyelinating disease. Usually the CSF shows a cellular response but no oligoclonal bands. Some have made an astonishing recovery after several months, and a few have then remained well for 25 to 30 years. Others have relapsed, and the subsequent clinical course was typical of MS.
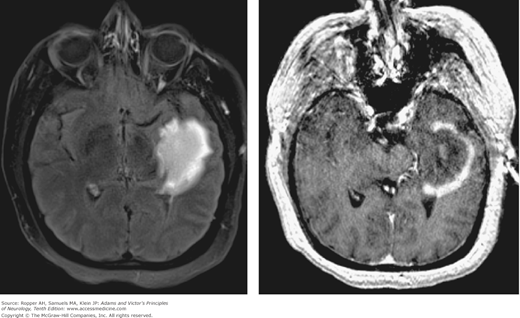
Among these cases are occurrences of large acute plaques with associated mass effect and enhancement that simulate a tumor on imaging (tumefactive MS, as described in the series by Kepes and shown in Fig. 36-2). The tumefactive lesion also occurs independently in cases of new or established disease that evolve in a manner that is more consistent with typical MS.
Related posts:
 Disorders of the Special Senses
Chapter 20. Delirium and Other Acute Confusional States
Chapter 24. Fatigue, Asthenia, Anxiety, and Depression
Disorders of the Special Senses
Chapter 20. Delirium and Other Acute Confusional States
Chapter 24. Fatigue, Asthenia, Anxiety, and Depression
 Chapter 28. Normal Development and Deviations in Development of the Nervous System
Chapter 28. Normal Development and Deviations in Development of the Nervous System
 Chapter 39. Degenerative Diseases of the Nervous System
Chapter 39. Degenerative Diseases of the Nervous System
 Chapter 38. Developmental Diseases of the Nervous System
Chapter 38. Developmental Diseases of the Nervous System
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


