Childhood Absence Epilepsy Evolving to Juvenile Myoclonic Epilepsy: Electroclinical and Genetic Features
Marco T. Medina*
Reyna M. Durón*
María E. Alonso‡
Charlotte Dravet§
Lourdes León||
Minerva Lopez-Ruiz¶
Ricardo Ramos-Ramirez¶
Ignacio Pascual Castroviejo†
Karen Weissbecker††
B. Westling†
Katerina Tanya Perez-Gosiengfiao**
Sonia Khan‡‡
Gregorio Pineda||
Ryoji Morita§§
Astrid Rasmussen‡
Jaime Ramos Peek||||
Sergio Cordova¶¶
Iris E. Martínez-Juarez†
Francisco Rubio-Donnadieu¶¶
Adriana Ochoa-Morales‡
Aurelio Jara-Prado‡
Julia N. Bailey†
Miyabi Tanaka†
Dongheng Bai†
Jesús Machado-Salas†
Antonio V. Delgado-Escueta†
*National Autonomous University of Honduras, Tegucigalpa, Honduras, †California Comprehensive Epilepsy Program, David Geffen School of Medicine at UCLA, Los Angeles, California
§Centre Saint-Paul-Hôpital Henri Gastaut, Marseille, France
||Hospital Angel Leaños, Guadalajara, Mexico
¶Neurology and Neurosurgery Unit, Mexico General Hospital, Mexico City, Mexico
††Tulane University Medical Center, New Orleans, Louisiana
**Department of Pediatrics, Autonoma University, Madrid, Spain
‡‡Neurosciences Department, Riyadh Armed Forces Hospital, Saudi Arabia
§§Laboratory for Neurogenetics, RIKEN Brain Science Institute, Saitama, Japan
||||Faculty of Medicine, Universidad Nacional Autónoma de México, University City, Mexico
†California Comprehensive Epilepsy Program, David Geffen School of Medicine at UCLA, Los Angeles, California, ¶Neurology and Neurosurgery Unit, Mexico General Hospital, Mexico City, Mexico
¶¶National Institute of Neurology and Neurosurgery, Mexico City, Mexico
‡Neurogenetics and Molecular Biology, Instituto Nacional de Neurología y Neurocirugía, Tlalpan, Mexico
†California Comprehensive Epilepsy Program, David Geffen School of Medicine at UCLA, Los Angeles, California
Introduction
The common idiopathic generalized epilepsies such as juvenile myoclonic epilepsy (JME), childhood absence (CAE), and grand mal (GM) on awakening are likely to be etiologically and genetically heterogenous (1,2,3,4,5,6,7). The same clinical phenotype may be caused by various single-gene defects or two or more genes interacting in epistasis and with environmental factors. JME begins around puberty with bilateral or unilateral, single or repetitive arrhythmic myoclonic jerks, which occur predominantly in shoulders and arms, usually after awakening or sleep deprivation, and with no associated disturbance of consciousness (8,9,10,11,12). Of JME patients 95% to 97% have grand mal, clonic–tonic–clonic or tonic–clonic convulsions. The interictal EEG shows diffuse 3.5- to 6-Hz polyspike-wave complexes and ictal EEG shows diffuse high-amplitude polyspikes during myoclonias (8,9,10,11,12,13,14,15). JME is most often inherited and sex distribution is reported to be equal or female preponderant, depending on the geographic and racial ethnic source (11,12,13,16,17,18,19,20).
In the original descriptions of JME, absences were considered a minor feature, as they occur in less than 30% of patients (10,12,14). When present, absences occurred rarely or in clusters with long periods of no attacks called spanioleptic. In recent studies, however, frequent absences numbered 1 to 200 per day, called pyknoleptic, have been recognized in JME (2,10,11,20,21,22). When the phenotypes of JME probands were analyzed, pyknoleptic absences were described in late childhood in 27%, in adolescence in 23%, and after 18 years in 5% (1,2). Durner et al. (4,5,23) and Greenberg et al. (6,7) claim that such absences and JME are inherited separately, which implies that separate genes are responsible for absences and myoclonia/grand mal.
Such pyknoleptic absence seizures have probably been responsible for our failure to isolate and identify JME genes. Families with subsyndromes of childhood absence epilepsy, such as CAE which evolves to JME or CAE with eyelid myoclonia and grand mal, or photogenic CAE with myoclonia and grand mal, are often mixed with families of classic JME in linkage mapping. Mixing these various subsyndromes together occur due to various reasons, one of which is the overlap of age at which absence epilepsy starts. Pyknoleptic absence in CAE usually begins between 2 and 12 years of age (2,9), while absences in JME have been reported between 8 and 18 years of age (14,24). Absences of the pyknoleptic variety with 3-Hz spike wave complexes have been reported in 4.6% (8,21), 5% (13), 7.5% (25), and up to 15% (9) of JME cases.
In this chapter, we describe the syndrome of childhood absence epilepsy persisting into adolescence with grand mal and myoclonias in 45 probands. We also describe the seizure phenotypes of 91 affected nonproband family members. We provide, in addition, results of a genome wide screen in a 100-member multiplex/multigenerational family ascertained through a proband with CAE evolving to JME. Results did not support linkage to seven known epilepsy loci, indicating this syndrome is a disorder distinct and separate from presently known epilepsy syndromes.
Methods
Patient and Family Database
Forty-five families (985 members) were ascertained through a proband with pyknoleptic childhood absence epilepsy that persisted with grand-mal and myoclonic seizures. Of the families, 29% (13/45) were multiplex/multigenerational, 27% (12/45) were multigenerational only, and 16% (7/45) were multiplex (Fig. 15-1). Of the families (13/45), 28% were simplex. Clinical diagnoses of seizure types and electrocephalographic diagnoses were independently verified by at least two epileptologists. Originally recruited for family studies in JME, these families were subsequently separated as a syndrome distinct from typical JME because of the onset of pyknoleptic absences during childhood (1,2,26). These families were evaluated from 1978 to 2004 in epilepsy clinics from Los Angeles (28 families), Mexico (10 families), Honduras (3 families), Saudi Arabia (3 families), and Spain (1 family). Two of the families evaluated in Los Angeles were originally from Iran and Australia, but were residing in the United States.
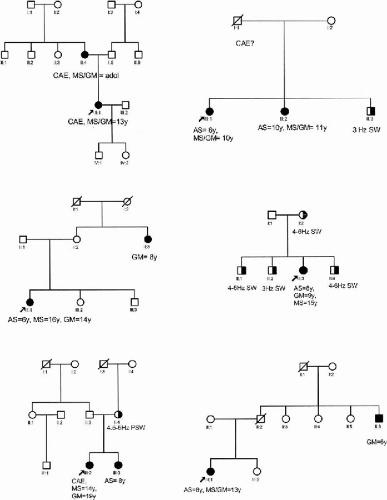 FIG. 15-1. Examples of multiplex and/or multigenerational families with CAE evolving to JME. AS, absences seizures; GM, grand mal; MS, myoclonic seizures; EM, eyelid myoclonia, SW, spike wave, TLE, temporal lobe epilepsy, FS, febrile seizures; PSW, polyspike-wave; EEG, electroencephalogram; JME, juvenile myoclonic epilepsy. |
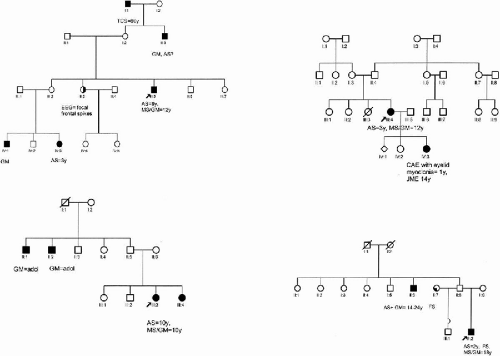 FIG. 15-1. (Contested) |
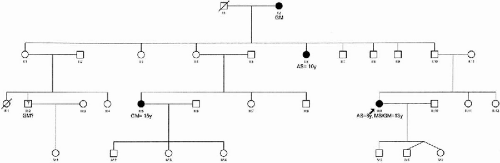 FIG. 15-1. (Contested) |
Inclusion criteria included (a) epilepsy that started in childhood with pyknoleptic absences (1–200 attacks per day) during 3-Hz spike-wave complexes; interictal EEG consisted of 3-Hz spike and wave and/or 3- to 6-Hz polyspike-wave complexes; (b) absences persisting beyond childhood; (c) myoclonic and tonic–clonic or clonic–tonic–clonic grand-mal seizures that started after absences, and (d) normal neurologic examination and brain imaging. Types, age at onset, and evolution of seizures through adolescence and adulthood were evaluated. The diagnosis of seizure types was based on the International League Against Epilepsy classifications of seizures and syndromes (27,28,29).
An extended pedigree was constructed for each family. All available affected relatives were interviewed and examined to determine their clinical diagnoses; EEGs were reviewed as available. Of the probands, 58 had neuroimaging studies—31% (14/45) had cerebral tomography scan, 18% (8/45) had brain magnetic resonance, 7% (3/45) had both tomography and magnetic resonance studies, and 18% (8/45) had positron emission tomography (PET). The number of relatives affected in nuclear and extended families, their sex, seizure types, and age at onset were registered.
Maternal or paternal transmission was determined by the pedigree findings. Exclusion criteria consisted of (a) progressive neurologic deterioration; (b) myoclonic absences; (c) typical or classic JME; (d) typical CAE that remits in adolescence, and (e) photogenic epilepsy with female preponderance. Of the patients (84%), 38 had follow-up to adolescence or adulthood.
One Large Family from Mexico Selected for Linkage Study
One large family whose pedigree structure and measures of informativeness (maximum expected LOD score) suggested strong power for linkage were selected to begin genome screening (Fig. 15-2). This large family was from Guadalajara, Mexico and had 100 members, 15 of whom were affected. This family was ascertained through a 15-year-old female proband with CAE evolving to JME. The proband and all nonproband members had 30- to 45-min electroencephalograms using disc electrodes applied according to the 10 to 20 system using colloidion or paste. Power test for this pedigree was simulated using the existing pedigree structure, affection status, and the program SLINK (Ott, 1989) (30). Assuming an autosomal dominant model, we simulated 1000 replicates. Based on quadratic interpolation, average maximum simulated LOD score was 4.5 ± 1.83.
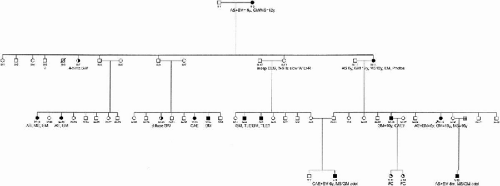 FIG. 15-2. Pedigree of a large family from Mexico selected for linkage analysis. See Fig. 15-1 for abbreviations. |
Loci Studied
Because of genetic linkage between classic JME and chromosome 6p12.1 (3,23,31,32,33,34), we initially screened the family from Guadalajara (Mexico) with chromosome 6 microsatellites. Subsequently, we performed a 10 to 20 cM genome screen using gel-based radiation microstellites from Research Genetics Inc. (Weber and May, 1989) (35) and fluorescent microstellite markers (ABI Prism genotyping system software, Perkin Elmer, Cyprus, CA). Data for reference families, primer sequences, allele sizes, and frequencies of each marker were obtained from the Genome Database or CEPH Database. Two-point and multipoint analysis used the LINKAGE Software Package (Version 5.21, 1998) (36,37). An autosomal dominant model with 70% penetrance and a disease frequency of 0.001 were used. We assumed no phenocopy, no mutation, and equal recombination between male and female.
Analysis of Familial Aggregation of Seizures and Epilepsy
For segregation analysis, each relative was examined separately for history of epilepsy and degree of relatedness. First-degree relatives were parents and siblings (brothers and sisters), second- degree relatives were uncles, aunts, nieces and nephews, and third-degree relatives included cousins. In addition, offsprings were analyzed separately from the other first-degree relatives. According to hospital and clinic records, CAE with or without grand mal accounts for 2% to 15% of all epilepsies (1,2,40). For risk calculations, we used the cumulative or lifetime prevalence of having epilepsy reported from the Rochester study (3%) (38,39), and considering that at least 2.2% of all epilepsies are CAE and 1.5% are JME (40), we estimated a prevalence of 0.0066% for CAE and a prevalence of 0.045% for JME in general population. Familial aggregation of both absences and myoclonic seizures were compared in the 32 multiplex-multigenerational pedigrees. Seizure types in nonproband members were compared with those of 92 classic JME families. Relative risk for every seizure type in families of CAE evolving to JME was compared with those of JME families.
Results
Probands
Thirty-one families (69%) were of Caucasoid ethnic origin, while fourteen (41%) were mixtures of European and American Indians. Females were preponderant among probands (29 females and 16 males or 1.8F:1M ratio). Figure 15-3 shows the age at onset of seizure types in probands. Childhood absence seizures were documented as the first seizure in all probands, starting at the average age of 6.9 ± 1.8 years (1 to 11 years) in 41 probands. In four other probands, parents could only describe absences as starting “during or before elementary school.” By the time probands were enrolled in family studies, they were either adolescents or adults (mean age 26.2 years with range 11 to 60 years), providing opportunity to observe the evolution of absences seizures that started during childhood. In 36 probands older than 20 years (80%), absences had persisted an average 22 ± 11 years (range 5 to 52 years).
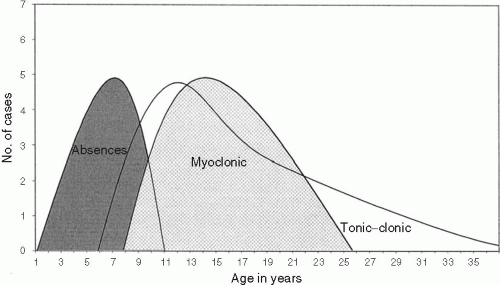 FIG. 15-3. Age at onset of seizures in 45 probands. |
Tonic–clonic seizures appeared for the first time at 12 years (range 2 to 37 years). Tonic–clonic convulsions were the chief persisting complaint in 21%. In probands older than 20 years, convulsive tonic–clonic seizures had been chronically recurring for 15 ± 6 years (range 3 to 23 years). All patients, except one, had at least one grand-mal seizure. The most common trigger factors for seizures were sleep deprivation in 33% (15/45), stress in 29% (13/45), light stimuli from TV, computers, and video games in 20% (9/45), and hyperventilation in 18% (8/45). Menses were reported as a trigger factor by 38% of female patients.
Myoclonias started at the mean age of 14 years (range 8 to 47 years) as sudden and involuntary jerks of shoulders and upper extremities, sometimes also affecting trunk, head, neck, and lower extremities. Drops due to severe myoclonic jerks or astatic seizures were not reported in probands. The most common description involved mild to moderate symmetrical, sudden lightning jerks of hands, forearms, and shoulders inward or upward, often causing the individual to drop items. Myoclonic seizures had persisted for 2 to 26 years (mean 12 years) in probands older than 20 years. Myoclonic seizures as the most prominent seizure type were observed in only six patients (13%). Myoclonic seizures preceded grand-mal convulsion in 18%. More often, they started simultaneously with tonic–clonic seizures in adolescence (40% of patients).
Neurologic Examination and Neuroimaging
Apart from seizures, all probands were otherwise neurologically normal. Neurologic examinations at enrollment and during the follow-up period remained normal. Five cases (11%) had prominent jaw jerk reflexes. Neither brain computed tomography (CT) nor magnetic resonance imaging (MRI) studies showed structural abnormalities among probands, except for one female who had an arteriovenous malformation (angioma venosum) in one frontal lobe, which was considered an incidental finding. Interestingly, two out of eight probands who underwent positron emission tomography (PET) were reported to have hypometabolism in one temporal lobe. One of them was the proband who had a frontal lobe arteriovenous malformation.
Electroencephalographs of Probands
Thirty probands (67%) had routine electroencephalograms (EEG), eight (17%) had EEG telemetry, and seven (16%) had video-EEG studies. Abnormalities were found in 95% of 37 probands (Fig. 15-4). The most common findings were 2- to 5-Hz single spike-and-slow wave complexes (78%) associated with absences. Of the probands, 54% had interictal 4- to 6-Hz polyspike-wave complexes, and 22% had interictal bursts of diffuse fast low amplitude 15- to 25-Hz rhythms in wakeful state. Background activity was normal in all cases. Bursts of 3- to 5-Hz single spike-wave and/or 4- to 6-Hz polyspike-wave were induced during hyperventilation in 29% of probands and during photostimulation in 22%. Myoclonias were associated with polyspikes or polyspike-wave complexes. Figures 15-5A-D show examples of these EEG traits. We recorded and videotaped myoclonic jerks associated with diffuse high-amplitude polyspikes and 4- to 5-Hz diffuse polyspike-wave complexes during video-EEG telemetry.
Related posts:
 Myoclonic Status in Nonprogressive Encephalopathies
Severe Myoclonic Epilepsy in Infancy: Dravet Syndrome
Idiopathic Myoclonic-Astatic Epilepsy of Early Childhood—Nosology Based on Electrophysiologic and Long-Term Follow-Up Study of Patients
Eyelid Myoclonia and Absence
Familial Adult Myoclonic Epilepsy (FAME)
Treatment of Myoclonic Epilepsies in Infancy and Early Childhood
Myoclonic Status in Nonprogressive Encephalopathies
Severe Myoclonic Epilepsy in Infancy: Dravet Syndrome
Idiopathic Myoclonic-Astatic Epilepsy of Early Childhood—Nosology Based on Electrophysiologic and Long-Term Follow-Up Study of Patients
Eyelid Myoclonia and Absence
Familial Adult Myoclonic Epilepsy (FAME)
Treatment of Myoclonic Epilepsies in Infancy and Early Childhood
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


