CHAPTER 140 Chordomas and Chondrosarcomas
Historical Background
Virchow first described small nodules along the clivus in 1846 and named them “ecchondrosis physaliphora” in 1857 because he hypothesized them to be of cartilaginous origin.1 The following year, Luschka, Henker, and Hasse described similar nodules. Concurrently, Muller postulated that ecchondrosis physaliphora was of notochordal rather than cartilaginous origin. In 1894, Ribbert coined the term “chordoma” and presented a case that supported Muller’s hypothesis. A year later, he experimentally induced chordomas in rabbits by perforating the anterior intervertebral ligament.1,2 In 1909, Harvey Cushing reported successful removal of a chordoma (initially described as a teratoma, case XVII) in a 35-year-old man (although the patient died during reoperation some 6 months later).3,4 Today, chordomas present the same challenges to neurosurgeons that they did 100 years ago; despite the remarkable advances made in operative and imaging techniques, the individual surgeon must often choose between the chance for cure and reduction of morbidity to achieve the greatest quality of life.
Natural History
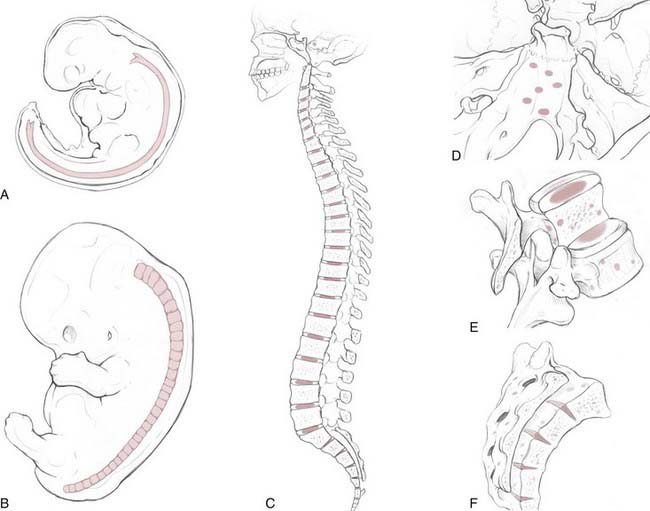
Chordomas are rare, slow-growing tumors that are believed to arise from rests of vestigial notochordal cells. They represent only 1% to 4% of primary bone tumors,5 with an age-adjusted incidence rate of 0.08 per 100,000 patient-years. There is a twofold greater incidence in males and a fivefold greater incidence in the white population.6 The peak incidence occurs in the eighth decade of life, and development of chordomas is quite rare before the age of 40. One recent, multi-institutional review suggested that anatomic location is evenly divided among the spheno-occipital region (32%), mobile spine (32.8%), and sacrum (29.2%).6 Older, often-cited reports, based on single-institution data, estimated that sacral chordomas constituted 50% of cases whereas 35% and 15% were cranial and spinal chordomas, respectively.5,7 Irrespective of the statistical distribution along the craniospinal axis, the anatomic diversity of skull base chordomas reflects the complex, serpentine course of the notochord within the clivus (Fig. 140-1). The clinical findings and symptomatology depend on the anatomic location of the tumor. Patients with cranial chordomas often have headaches, neck pain, cranial neuropathies, and even endocrine abnormalities, whereas those with spinal chordomas have back pain, pathologic fractures, myelopathy, and radiculopathy. Sacral chordomas tend to be quite large at the time of diagnosis because of the insidious onset of symptoms such as pain and bowel or bladder dysfunction.8
Overall, 5- and 10-year survival rates are 67.6% and 39.9%, respectively.6 Local recurrence is the most important predictor of mortality in patients with sacral chordomas. Distant metastases to the lung, liver, and bone can occur with chordomas, but they generally take place quite late in the disease process and, as a result, probably contribute little to overall mortality.
The notochord is an evolutionarily conserved structure (hence, the phylum Chordata) that arises early in embryonic development from the dorsal organizer region. It plays a critical role in many aspects of development, such as determining the dorsal-ventral axis of the neural tube (through secretion of the sonic hedgehog homologue [SHH] gene product), left-right asymmetries, and arterial-versus-venous fates of early blood vessels. The notochord is made up of highly vacuolated cells (the physaliphorous cells) that contain a hydrated protein matrix and are surrounded by a thick basement membrane. The protein matrix absorbs water, which allows the cells to swell and exert pressure on the basement membrane. In this fashion, the notochord provides structural support to the developing embryo. In animals with a bony spine, the notochord involutes as the adjacent somites form the vertebral bodies. Some notochordal cells persist and form the nucleus pulposus of the intervertebral disks, whereas others survive as “rests” within the clivus, sacrum, and vertebrae.9,10 The relationship between these cellular rests and the development of later chordomas is complex. Along the dorsal clivus, for example, approximately 2% of the population, based on autopsies (Ribbert’s theory, see Bailey and Bagdasar1), have benign growths termed ecchordosis physaliphora (i.e., the ecchondrosis of Virchow). Rarely, they can grow and become symptomatic giant ecchordoses or intradural benign chordomas.11,12 Similarly, benign notochordal tumor is a recently recognized entity found within the clivus (11.5% of autopsies) and vertebral bodies (14% of autopsies) that can expand and become a classic chordoma.13,14 These tumors are immunohistochemically distinct from fetal notochord rests.13 However, whether all classic chordomas arise from benign tumor precursors is unknown.
Radiography
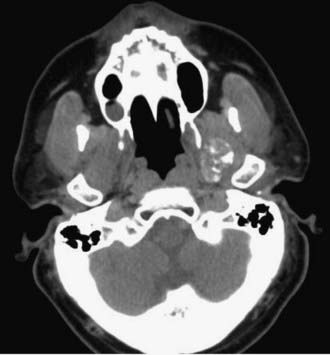
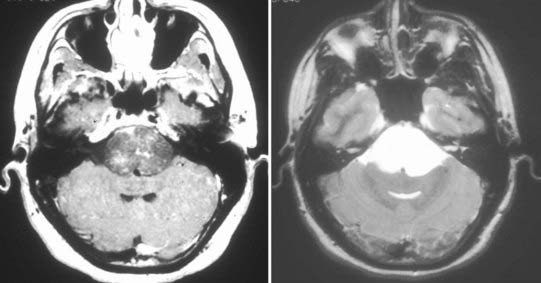
Chordomas and chondrosarcomas are indistinguishable from one another on radiographic examination, except for the fact that chondrosarcomas tend to arise from a paramedian location and chordomas are nearly always found along or near the midline.15–18 One caveat in this regard is that chordomas can distort their midline origin by becoming quite large before they are detected and growing asymmetrically in one direction or another. This asymmetric growth may extend into the nasopharynx, orbits, or sella or along the cranial nerves, with dorsal deflection of the brainstem. Although chondrosarcomas typically arise from the skull base, they can also arise from the meninges. On non–contrast-enhanced computed tomography (CT), both chordomas and chondrosarcomas are slightly hyperdense masses with variable amounts of calcification (Fig. 140-2). Bone windows often demonstrate significant bony erosion. For chordomas, the erosion is predominantly along the clivus and petrous apex. Tumors of both types show variable enhancement on contrast-enhanced CT. Chordomas and chondrosarcomas are both isointense to hypointense on T1-weighted magnetic resonance imaging (MRI), again with variable enhancement. On T2-weighted MRI these tumors are hyperintense, but they may have some heterogeneity in signal intensity because of calcifications and bony sequestrations (Fig. 140-3). Benign ecchordosis physaliphora is radiographically similar but does not show enhancement.19,20
Pathology
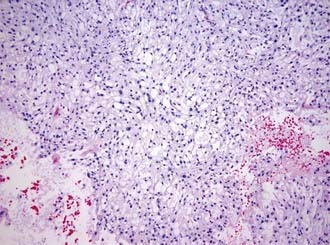
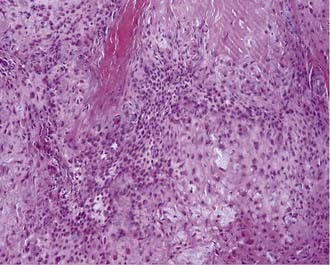
Macroscopically, chordomas and chondrosarcomas are nodular, exophytic masses with a smooth whitish or occasionally reddish appearance.1,21 They often display a fibrous outer layer that is contiguous with the fibrous septations found within the tumor. This pattern of growth gives the tumor a lobulated appearance in cross section. These tumors lack a thick capsule. Internally, the tumors are soft, gray, and translucent but generally lack significant areas of necrosis. In addition to their local bony destruction, these tumors will extend out into adjacent cancellous bone, thus making oncologic excision difficult if not impossible. Microscopically, chordomas can be divided into three main histochemical types: conventional, chondroid, and dedifferentiated. Conventional chordomas are composed of sheets or cords of cells with eosinophilic cytoplasm embedded in a mucinous matrix. The appearance of these cells can range from having a homogeneous cytoplasm completely lacking in vacuoles to a cytoplasm markedly engorged with vacuoles, the so-called physaliphorous or bubble-bearing cells (Fig. 140-4). Differentiation into cartilaginous or, occasionally, osseous-appearing cells may occur focally or throughout the tumor and, if extensive, distinguishes the chondroid chordoma subtype from conventional chordomas. Rarely, chordomas undergo malignant transformation, which gives rise to dedifferentiated chordomas. In this subtype there is a predominant sarcomatous appearance with malignant spindle cells, increased atypia, and increased mitotic index. Immunohistochemically, chordomas are reactive for epithelial membrane antigen, cytokeratins, α-fetoprotein, and rarely, S-100.
Chondrosarcomas can also be divided into three subtypes: classic or conventional, mesenchymal, and dedifferentiated. Conventional chondrosarcomas are composed of large, often multinucleated cells within a cartilaginous matrix (Fig. 140-5). Their pathology has three histologic grades. The degree of atypia, number of mitoses, nuclear-to-cytoplasmic ratio, and amount of cartilaginous matrix correlate with the aggressiveness of the tumor, with grade III being the most aggressive. Mesenchymal chondrosarcomas have islands of cartilage mixed with primitive mesenchymal cells, and undifferentiated tumors have a marked sarcomatous appearance. Both mesenchymal and undifferentiated chondrosarcomas have a more aggressive course. Immunohistochemically, chondrosarcomas lack the epithelial antigens expressed by chordomas despite a histologic appearance similar to that of chondroid chondromas.

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


