Chromosomal Abnormalities
Renzo Guerrini
Agatino Battaglia
Romeo Carrozzo
Giuseppe Gobbi
Elena Parrini
Tiziano Pramparo
Orsatta Zuffardi
Introduction
About 30 years ago, chromosomal abnormalities, as detectable by classic cytogenetics, were estimated to cause approximately 6% of central nervous system (CNS) malformations.31 Today, the use of current techniques such as high-resolution chromosome banding, fluorescent in situ hybridization (FISH), and molecular genetics would certainly increase this percentage; it was estimated recently that genome-wide molecular cytogenetics techniques (array-CGH) can detect cryptic deletions/duplications in no less than 20% of individuals with mental retardation and dysmorphic features but apparently normal karyotype (Fig. 1).139 It is likely that array-CGH at higher resolution will increase this percentage even further.
Most syndromes associated with chromosomal abnormalities are caused by duplications (with duplication of a segment of chromosome), deletions (absence of a segment), or breakpoint disruptions (only one or a few genes are disrupted). Virtually all known chromosomal abnormalities lead to anatomic and functional impairment of the CNS and, especially those involving autosomes, are accompanied by mental retardation. Tharapel and Summit155 found 6.2% of cases of mental retardation to be associated with chromosomal abnormalities, as contrasted with the 0.7% rate found in controls.
The risk of epilepsy is greater in individuals with chromosomopathy than in the general population.74 However, chromosomal abnormalities do not represent a frequent cause of epilepsy.78
Epilepsy as a complication of a chromosomal abnormality syndrome should be considered in a twofold perspective. First, symptomatic epilepsies have a high probability of causing intractable seizures, leading to further disability in these patients. Severe epilepsy may significantly reduce a low-IQ patient’s potential for autonomy in everyday life.64 Second, when seizures are the main manifestation of a chromosomal disorder or present with peculiar electroclinical patterns, it is important to establish whether these associations result from structural CNS abnormalities caused by the chromosomal changes or from the effect of loci that specifically affect seizure susceptibility. Careful attention to chromosomal abnormalities associated with seizures, together with detailed analysis of electroclinical patterns, may help to detect specific genes affecting seizure susceptibility.2,142 It seems likely that the association between cryptic deletions/duplications and epilepsy will improve our ability to clone new critical genes.
Only those chromosomal abnormalities of special importance for their high frequency or association with epilepsy are considered in this chapter.
Standard Cytogenetic and Molecular Techniques for Diagnostics
Standard Karyotype and High-Resolution Chromosome Analysis
The karyotype of an individual can be ascertained from readily accessible somatic cells, such as peripheral blood lymphocytes or skin fibroblasts. Each chromosome can be identified by special staining techniques, using fluorescent dyes or Giemsa staining after treatment with a proteolytic enzyme (trypsin). A typical standard karyotype from metaphase cells contains 400 to 550 bands per haploid genome. “High-resolution” chromosome analysis makes it possible to visualize up to 2,000 bands, although a high-resolution chromosome analysis of standard quality allows the detection of about 850 bands. Given the greatly increased amount of work, the difficulty of the analysis, and the cost, high-resolution chromosome analysis should not be considered as a routine cytogenetic assay.
Molecular Karyotyping with Array-CGH
Several techniques defined as molecular cytogenetics have, over the course of the last 10 years, improved the resolution capability of the chromosome banding. The latest technology is the array-CGH, which represents a powerful modification of classical comparative genomic hybridization (CGH) based on metaphases. Using CGH, the DNA of a patient (to be tested) and the DNA of a control are labeled with green and red fluorochromes, respectively, and hybridized on metaphase spreads. The images of both fluorescence signals are captured, and the ratio of DNA intensities quantified using a dedicated software along each chromosome. The chromosome regions represented in the same amount in both DNAs (patient and control) appear yellow, the deleted regions appear red, and the duplicated regions appear green. The difficulty of obtaining sufficiently long metaphases limits the resolution power of this technique to a maximum of 3 Mb. Array-CGH technology replaces the use of chromosome spreads with clones or oligomers (targets) that cover the entire genome, and its resolution power depends only on the amount of targets and their distribution. Recently, microarrays using bacterial artificial chromosome (BAC) clones separated by 100 Kb have been used. Other platforms, using oligomers as targets, have a higher resolution that theoretically can reach up to a few hundreds of base pairs. The use of these arrays might allow the detection of deletions and duplications below the size of 1 Mb and precisely define their
breakpoints (Fig. 1). This technology has also revealed that the human genome may harbor deletions and duplications, having an average size of 2 Mb and being devoid of phenotypic effects, which may play a role in multifactorial diseases. It seems obvious that microarray-CGH will have a crucial role in pre- and postnatal cytogenetics as well as in oncology; its potential in the etiologic diagnosis of epilepsy associated with mental disability and dysmorphic features is extremely high.
breakpoints (Fig. 1). This technology has also revealed that the human genome may harbor deletions and duplications, having an average size of 2 Mb and being devoid of phenotypic effects, which may play a role in multifactorial diseases. It seems obvious that microarray-CGH will have a crucial role in pre- and postnatal cytogenetics as well as in oncology; its potential in the etiologic diagnosis of epilepsy associated with mental disability and dysmorphic features is extremely high.
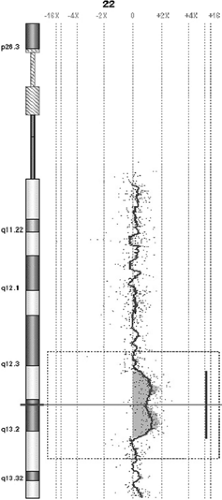 FIGURE 1. Eight-year-old girl with early-onset focal epilepsy and dysmorphic features. 6.9 Mb duplication at 22q13.1q13.2 detected through oligo-array-CGH at 75 kb resolution; log2 ratio of signals intensity is increased in the duplication region where red signals are located more proximal to the +2x line than are the remaining ones. This interstitial duplication had not been detected at conventional karyotyping nor, obviously, after FISH with subtelomeric probes. (See color insert.) |
Fluorescent in Situ Hybridization
The hybridization of a labeled DNA probe for spreading metaphase chromosomes allows the detection of small deletions, duplications, or cryptic translocations involving the chromosomal segment complementary to a probe labeled with a fluorochrome. The probe signal can be visualized by fluorescence microscopy. Double or multiple hybridizations can be carried out at the same time using different fluorochromes. Although this is a sensitive technique, it allows the investigation of only a few loci in one experiment, thus it can be requested when the clinician already has a specific clinical suspicion known to be associated with the deletion/duplication of that gene/region (i.e., Angelman syndrome and 15q11–q13 deletion).
Southern Blot Analysis
Differences in the DNA sequences at the same locus in different individuals can be detected by restriction enzyme digestion and Southern blot analysis. The diversity in DNA sequences among different individuals will result in either the introduction or loss of specific restriction sites, thereby causing DNA fragments of different sizes. The DNA is subsequently electrophoresed on an agarose gel, denatured with alkali, transferred to a nylon filter, and hybridized with a radiolabeled probe. The resulting X-ray film permits a comparison among the segments of DNA from different individuals.
Polymerase Chain Reaction
Polymerase chain reaction (PCR) allows the amplification of DNA fragments of 100 to 2,000 bp in length. The reaction is carried out in the presence of oligonucleotides flanking the region of interest, free deossinucleotides, template DNA, thermostable DNA polymerase from Thermophilus aquaticus (TAQ), and an appropriate saline buffer. The reaction requires 25 to 35 cycles of denaturing, annealing, and synthesizing DNA strands complementary to the template, thus allowing an exponential increase in the amount of target DNA for each cycle. The PCR product can be visualized on an agarose gel stained with ethidium bromide or labeled with a radioisotope, run on a polyacrylamide gel, and detected after exposure of the gel to an X-ray film.
Main Chromosomal Abnormalities Associated with Epilepsy
Trisomy 21 (Down Syndrome)
General Clinical Findings
Down syndrome (DS) has an approximate incidence of 1 in 650 births.144 In 95% of cases, the cause is a nondisjunction of chromosomes 21 during meiosis; in about 4% of cases, there is an unbalanced translocation. Approximately 1% of patients are mosaics and show a less severe phenotype. The region that, if triplicated, results in the typical phenotype maps to 21q22.3.41 The risk of carrying a child with DS increases with increasing maternal age and with very young maternal age.79
The main clinical features of this condition include growth retardation, mental retardation of variable degree, hypotonia, flat facies, brachycephaly with flat occiput, upward-slanting eyes, epicanthal folds, small ears, speckling of the iris (Brushfields spots), simian crease, and hypogonadism. About 40% of patients have congenital heart disease. There is a marked risk of leukemia. All individuals above the age of 35 years show features of Alzheimer disease.
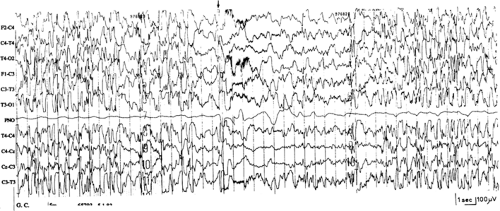 FIGURE 2. Five-month-old girl with Down syndrome and infantile spasms. Typical and symmetrical hypsarrhythmia that is interrupted after a spasm (arrow) initiating a cluster of spasms (not shown). Note rapid rebuilding of hypsarrhythmia after the spasm. |
Epilepsy
Epilepsy in DS has been extensively investigated.45,60,66,83,135,147 The incidence of epilepsy in children with DS was estimated to be 1.4%.153 However, the overall prevalence of epilepsy increases with age, reaching 12.2% in patients over 35 years
of age.159 About 40% of patients have seizures since the first year of life, and another 40% experience their first seizure during the third decade.116 This trend seems to be related to the early medical complications of DS, such as hypoxic ischemic encephalopathy and congenital heart disease,129 and the development of neuropathologic changes typical of Alzheimer disease36 have a specific etiology, most often related to common medical complications of DS such as hypoxic–ischemic perinatal suffering, hypoxia from congenital heart disease, or infection. Febrile seizures have a low frequency (0.9%) in comparison with the general population.60,93,129,153 When only cases without known causes were considered, 2.5% of patients presented with seizures, a frequency still greater than that in the general population. Although many patients in both groups had their first seizure in the first year of life, seizures occurred significantly later in infants without a known etiology. Generalized tonic–clonic (GTC) seizures predominated in both the known and unknown etiology groups. Myoclonic seizures and infantile spasms with hypsarrhythmia were also common, the latter predominating in the unknown etiology group. Partial seizures usually occurred in patients with an identifiable etiology. Prognosis for recurrent seizures varied with the etiology. Patients with seizures related to cardiovascular disease were usually well controlled by anticonvulsants. Neonates with hypoxic–ischemic injury had poor outcomes. None of the patients who developed seizures as a result of CNS infection had persistent attacks. Patients with idiopathic seizures had generally good outcomes. The authors concluded that children with DS have an increased susceptibility to seizures early in life, and superimposed systemic illness increased the risk.
of age.159 About 40% of patients have seizures since the first year of life, and another 40% experience their first seizure during the third decade.116 This trend seems to be related to the early medical complications of DS, such as hypoxic ischemic encephalopathy and congenital heart disease,129 and the development of neuropathologic changes typical of Alzheimer disease36 have a specific etiology, most often related to common medical complications of DS such as hypoxic–ischemic perinatal suffering, hypoxia from congenital heart disease, or infection. Febrile seizures have a low frequency (0.9%) in comparison with the general population.60,93,129,153 When only cases without known causes were considered, 2.5% of patients presented with seizures, a frequency still greater than that in the general population. Although many patients in both groups had their first seizure in the first year of life, seizures occurred significantly later in infants without a known etiology. Generalized tonic–clonic (GTC) seizures predominated in both the known and unknown etiology groups. Myoclonic seizures and infantile spasms with hypsarrhythmia were also common, the latter predominating in the unknown etiology group. Partial seizures usually occurred in patients with an identifiable etiology. Prognosis for recurrent seizures varied with the etiology. Patients with seizures related to cardiovascular disease were usually well controlled by anticonvulsants. Neonates with hypoxic–ischemic injury had poor outcomes. None of the patients who developed seizures as a result of CNS infection had persistent attacks. Patients with idiopathic seizures had generally good outcomes. The authors concluded that children with DS have an increased susceptibility to seizures early in life, and superimposed systemic illness increased the risk.
In a review of studies in which population estimates were possible, infantile spasms occurred in 0.6% to 13% of children with DS, representing 4.5% to 47% of all seizures (reviewed by Stafstrom and Konkol146). Infantile spasms probably represent the most common seizure type in DS and, in most children, they appear without any evidence of additional brain damage.60,113,141,146 Prognosis is usually good with regard to seizure control. Remission was obtained on conventional antiepileptic drugs (AEDs), adrenocorticotropic hormone (ACTH), or steroids, without relapse of seizures60,113,146 or with later onset of pharmacologically controllable, age-related generalized seizure disorders141 (Fig. 2). Conversely, spasms proved to be resistant, progressing to other forms of intractable epilepsy in most of the patients who suffered hypoxic insults.60,146
The occurrence of reflex seizures in the context of startle epilepsy is particularly frequent (Fig. 3).54,66,115,130 Age at seizure onset is variable, and there is usually no evidence for etiologic factors other than DS. Most of the patients described presented “pure” forms of reflex epilepsies, with almost all seizures being precipitated by acoustic or tactile stimuli. However, the epileptic syndrome varied in type and severity, with a predominance of Lennox-Gastaut–type patterns.66 The mechanism leading a high incidence of reflex phenomena in DS patients is unclear. A central deficit of inhibition of afferent stimuli has been proposed.29
Lennox-Gastaut syndrome (LGS) has been well documented in DS, but is not frequent. Quite characteristic is its de novo appearance at a mean age of 10 years, not preceded by other forms of epilepsy.60,66
Only a few studies have analyzed adult or late-onset epilepsy in DS, despite its high frequency.90,159,164 Although in most late-onset epilepsies seizures appeared infrequently but were intractable,60 no precise indications on seizure severity are available. Late-onset generalized epileptic myoclonus has also been reported.53
Neuropathology and Neurogenetic Basis of Seizures
Brain weight is in the lower normal range, and the gyral pattern is simplified.39 A narrow superior temporal gyrus is observed in about 50% of patients. The insula is exposed because of hypoplasia of the inferior frontal gyrus.86 Changes typical of Alzheimer disease appear above the age of 30 years.51,64 Several cytoarchitectonic abnormalities have been found from birth, including a 20% to 50% decrease in the number of small granule cells, mainly inhibitory γ-aminobutyric acid (GABA)ergic interneurons, with lower neuronal density, abnormal neuronal distribution, and dysgenesis of dendritic spines.18,81,98,127,166 The relation among these structural abnormalities and increased susceptibility to certain seizure types, particularly infantile spasms and reflex seizures, is poorly understood.
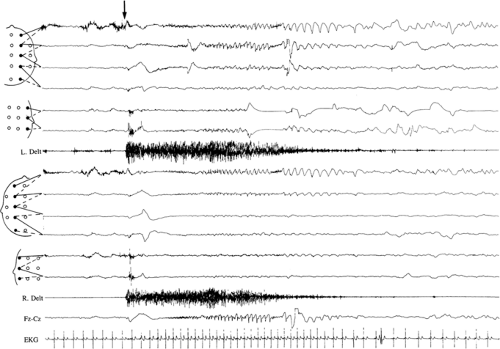 FIGURE 3. Twenty-year-old male Down syndrome patient with startle epilepsy. A sudden, unexpected noise induces a tonic seizure lasting 15 seconds, accompanied by tachycardia; the patient, who is sitting on the bed, is projected violently backward. Tonic contraction, visible on the deltoid muscles (R and L Delt), is accompanied on the EEG by a decremental event, followed by low-voltage fast activity, with increasing amplitude and decreasing frequency, well recognizable over the vertex and frontocentral electrodes, more on the right. |
Diagnostic Evaluation
Most patients have a free trisomy 21, detectable on standard chromosome analysis. In such circumstances, chromosome study in the parents is not indicated. Parents should be given a 1% recurrence risk, based on the calculation of the recurrence of DS in a sibship. This slightly higher recurrence risk, when compared to the expectation based on the maternal age is likely due to a germinal mosaicism present in a few parents or to a general failure in the chromosomal disjunction mechanisms normally occurring at meiosis.110
If DS results from a translocation, this usually occurs between acrocentric chromosomes, termed a robertsonian translocation. In such circumstances, one of the parents might be a balanced carrier of the translocation, and a chromosomal study is strongly indicated for both parents. The most common robertsonian translocation found in healthy carriers involves chromosomes 14 and 21. Virtually all the (21;21) translocations are de novo. The involvement of chromosome 21 in translocations with nonacrocentric chromosomes101 is very rare. The calculation of the recurrence risk for DS in the carriers of robertsonian translocation is based on the gender of the transmitting parent and the chromosome involved.
Fragile-X Syndrome
General Clinical Findings
Fragile-X syndrome has an approximate incidence of 1 in 1,500 males163 and is the most common chromosomal abnormality associated with heritable mental retardation. Both genders may be affected, but the phenotype is notably more severe in males. It is estimated that 1 in 1,000 females are carriers.23 The X-chromosome of patients affected with the syndrome shows a “fragile” site at Xq27.3 when cells are grown in a folic-deprived medium. The condition results from a dynamic mutation in heritable unstable DNA,124 because of variation in the copy number of a trinucleotide repeat p(CCG)n within the FMR1 gene. This fragile site is termed FRAXA. The disorder has an unusual mode of inheritance. About 20% of males who carry the mutation are clinically and cytogenetically normal (normal transmitting males). About 30% of female carriers have mental impairment. Transmission is consistent with an X-linked semidominant condition. The main clinical features include moderate mental retardation, poor language, hypotonia, growth retardation, macrocephaly, prominent forehead, long, narrow facies, large ears, macroorchidism, pectus excavatum, a floppy mitral valve, and hyperactive or autistic behavior.50,165
Epilepsy
The prevalence of epileptic seizures is approximately 25%.167 Seizures usually appear before the age of 15 and tend to disappear during the second decade of life.65,167 In most patients, seizures are fairly rare or are controllable with simple drug regimens.59,65,105,167 The most frequently mentioned seizure types are generalized tonic–clonic seizures.50,165 However, no specific epilepsy syndrome is observed.65 Background electroencephalographic (EEG) activity is slow.105,167 An EEG pattern of midtemporal spikes, possibly age related, similar to the waveform of benign rolandic epilepsy, has been described in some affected males, with or without seizures.104 This EEG pattern has been confirmed by other investigators, but only in a minority of cases.60,167 The fragile-X locus has been excluded as the candidate gene for benign rolandic epilepsy.118
Neuropathology and Neurogenetic Basis of Seizures
The variety in seizure types in the fragile-X syndrome may reflect extreme clinical polymorphism as a consequence of the variability of amplification of the trinucleotide repeat among patients.52 A somatic variation in DNA amplification during development is also possible.168 Little is known of the neuropathology. Anomalous synapses and abnormal development of dendritic spines have been observed using Golgi impregnation.129,167 Neuroimaging studies have shown hypoplasia of the cerebellar vermis121 and periventricular heterotopia in rare patients.103
Diagnostic Evaluation
The fragile site at Xq27.3 is not expressed in cell culture media employed for standard karyotypes. The induction of the fragile site in stimulated lymphocytes can be achieved using culture media free of folic acid and thymidine. The fragile site is detected only in a proportion of metaphase cells, with most fragile-X males expressing their fragile site in only 5% to 50% of their lymphocytes. Normal transmitting males usually do not express the fragile site. Fragile-X females with mental impairment express their fragile site, but in a lower proportion of cells than do fragile-X males. The majority of female carriers do not express the fragile site, but some of them might show an expression in 5% or less of their cells. The fragile-X syndrome is due to the expansion of a p(CGG)n repeat within the 5′ untranslated region of the FMR1 gene. Normal chromosomes are polymorphic, containing 6 to 52 copies of this repeat, whereas males with more than 200 copies have the disease (full mutation). Normal transmitting males have 50 to 200 copies (premutation). Females bearing the premutation are phenotypically normal and do not express the fragile site. Females with more than 200 copies may be normal or mentally retarded and can show facial features of fragile-X syndrome. Premutations tend to expand when transmitted to offspring through female carriers, and the risk of expansion to a full mutation correlates with the size of the premutation.52,73 In association with the full mutation, the promoter region of the FMR1 gene is hypermethylated, and the transcription of the gene is repressed.112,160 DNA studies have improved the accuracy of testing for fragile-X syndrome. By looking at the size of the trinucleotide repeat segment, as well as the methylation status of the FMR1 gene, the genotype can be determined for both affected individuals and suspected carriers. Two main approaches are used: Polymerase chain reaction (PCR) and Southern blot analysis. PCR analysis utilizes flanking primers to amplify a fragment of DNA spanning the repeat region. Thus, the sizes of the PCR products are indicative of the approximate number of repeats present in each allele of the individual being tested. The efficiency of the PCR reaction is inversely related to the number of CGG repeats, so that large mutations are more difficult to analyze and may fail to yield a detectable product in the PCR assay. An additional limitation to the PCR approach is due to the fact that it provides no information about FMR1 methylation. On the other hand, PCR analysis permits accurate sizing of alleles in the normal and premutation size ranges. FMR1 analysis by Southern blotting allows both size of the repeat segment and methylation status to be assayed simultaneously. A methylation-sensitive restriction enzyme that fails to cleave methylated sites is used to distinguish between methylated and unmethylated alleles. Southern blot analysis is more labor intensive than PCR and requires larger quantities of genomic DNA. Southern blot accurately detects alleles in all size ranges, but precise sizing is not possible. Many laboratories can use both methods, and choose the type of analysis that is most appropriate to the circumstances. A few patients with fragile-X harboring deletions in the FMR1 have been described.117,152
Two additional fragile sites reside in the distal Xq, termed FRAXE and FRAXF. Although FRAXF is not clearly associated with a specific phenotype, those with FRAXE show mental impairment, usually milder than that observed in patients with FRAXA mutations. Because no clinical overlap occurs between patients with mutations at the FRAXA and the FRAXE loci, other than mental retardation, the details about the diagnostic procedures for identifying FRAXE will not be discussed here.
Del 4p (Wolf-Hirschhorn) Syndrome
General Clinical Findings and Genetic Background
Wolf-Hirschhorn syndrome (WHS) is a multiple congenital anomalies/mental retardation syndrome34 with a frequency of 1 in 50,000 births and a female:male predilection of 2:1.57,96 It is caused by partial loss of material from the distal portion of the short arm of chromosome 4. The minimal deleted segment causing the phenotype is 4p16.3.96 Although about 75% of patients have a de novo deletion96 of preferential paternal origin,38,157 about 12% have an unusual chromosome abnormality (such as ring 4), and about 13% have a deletion of 4p16 resulting from an inherited unbalanced chromosome rearrangement from a parent with a balanced rearrangement. The main features of monosomy 4p are the typical “Greek warrior helmet appearance of the nose,” microcephaly, pre- and postnatal growth delay, congenital hypotonia, severe mental retardation, and seizures.13 In at least one-third of cases, death occurs during the first year of life because of severe systemic malformations, cardiac failure, or pulmonary infection.
Epilepsy
Seizures constitute a major medical concern during the first years of life, and occur in 50% to 100% of patients.10,13,32,40,68,149 Seizure onset usually occurs within the first 2 years of life, with a peak incidence around 9 to 10 months of age. These seizures may be clonic or tonic, unilateral with or without secondary generalization, or generalized tonic–clonic from the onset.5,8,9,10,11,12,13,47,108,119 They are frequently triggered by fever, may be prolonged, and often occur in clusters.5,11,12,136,171 More than 50% of the patients experience early unilateral or generalized clonic or tonic–clonic status epilepticus (SE), despite adequate treatment.13 Two-thirds of the subjects develop, between 1 and 5 years of age,
atypical absences, often accompanied by a mild myoclonic component, mainly involving the eyelids and axorizomelic muscles.5,11,12,13,14,136 Such episodes are accompanied by generalized slow spike-and-wave complexes. The interictal EEG shows high-amplitude, fast spikes, polyspikes, and wave complexes over the posterior third of the head on eye closure. Other seizure types have been described in a minority of patients (Fig. 4).80,171
atypical absences, often accompanied by a mild myoclonic component, mainly involving the eyelids and axorizomelic muscles.5,11,12,13,14,136 Such episodes are accompanied by generalized slow spike-and-wave complexes. The interictal EEG shows high-amplitude, fast spikes, polyspikes, and wave complexes over the posterior third of the head on eye closure. Other seizure types have been described in a minority of patients (Fig. 4).80,171
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


