Chapter 41 Circadian Disorders of the Sleep–Wake Cycle
Introduction
The sleep–wake cycle is generated by a complex interaction of endogenous circadian and sleep homeostatic (need for sleep increases as a function of prior wakefulness) processes, as well as social and environmental factors. Physiological sleepiness and alertness not only varies with prior waking duration, but also exhibits circadian variation. In humans, daily variation in physiological sleep tendency reveals a biphasic circadian rhythm of wake and sleep propensity,1,2 with a midday increase in sleep tendency occurring around 2 to 4 PM, followed by a robust decrease in sleep tendency and increase in alertness that lasts through the early to midevening hours. A primary role of the circadian clock is to promote wakefulness during the day, and thus to facilitate the consolidation of sleep during the nighttime hours.1,3–5 In humans, this interaction between circadian and homeostatic processes results in approximately 16 hours of wakefulness and 8 hours of nocturnal sleep.
The timing and duration of the sleep–wake cycle depends upon the synchronization of the endogenous circadian clock with the external physical light and dark cycle and social or professional demands. Circadian rhythm sleep disorders (CRSD) arise when there is disruption of this internal timing mechanism or a misalignment between the timing of the circadian clock and the 24-hour social and physical environments. These dyssynchronous states fall into two categories: (1) the terrestrial light–dark (LD) cycle may change relative to circadian timekeeping (shift work disorder and jet lag), or (2) circadian timekeeping may change relative to the terrestrial LD cycle (delayed sleep phase disorder [DSPD], advanced sleep phase disorder [ASPD], free-running disorder (FRD) and irregular sleep–wake rhythm). The first circumstance occurs in the presence of a normal circadian timekeeping system and is generally self-limited or resolves with environmental change. The latter circumstance is believed to occur because of chronic alterations in the circadian system that result in the inability of the circadian pacemaker to achieve a conventional phase relation with the external world. This chapter will focus on the second group of disorders including DSPD, ASPD, FRD, and irregular sleep–wake rhythm, whereas shift-work sleep disorder and jet lag are covered extensively in Section 9, Chapter 71. Because circadian variation in wakefulness and sleep propensity are the most apparent of the many behavioral and physiologic outputs of the circadian pacemaker, it is not surprising that the most apparent circadian rhythm disorders involve the sleep–wake cycle.6
Entrainment of Circadian Rhythms
The suprachiasmatic nucleus (SCN), located in the anterior hypothalamus, is the central pacemaker responsible for the generation of circadian rhythmicity in mammals.7 Animals and humans removed from the external LD cycle and other time cues (zeitgebers) exhibit a self-sustaining cycle of sleep and wakefulness as well as many other physiologic and hormonal rhythms.
The endogenous frequency of this cycle of oscillation or free-running period is largely genetically determined.8 The basic molecular mechanism by which SCN neurons generate and maintain a self-sustaining rhythm is via an autoregulatory feedback loop in which oscillating circadian gene products regulate their own expression through a complex system of transcription, translation, and posttranslational processes.9 In the mouse10 and hamster,11 genes have been identified that lengthen10 and shorten11 the free-running period. The mammalian circadian period is generally slightly longer than 24 hours in diurnal animals, and slightly shorter than 24 hours in nocturnal animals. In humans, the average circadian period has been estimated to be approximately 24.18 hours12; therefore, it must be synchronized or entrained on a regular basis to the 24-hour terrestrial day by external influences.
Entrainment by Light
Light is the major external time cue in mammals. Light reaches the SCN by afferent projections from the retina via the retinohypothalamic tract.7 Evidence indicates that the primary circadian photoreceptors are the melanopsin-containing retinal ganglion cells, which in turn send photic information via projections to the SCN.13,14
Although circadian rhythms can be entrained to LD cycles that are not exactly 24 hours long, entrainment is restricted to cycles with periods that are close to 24 hours long.15 The range of entrainment can vary from species to species and is dependent on the experimental conditions (e.g., intensity of light–dark cycle, whether the period of the light–dark cycle is changed gradually or rapidly), but in general, animals do not entrain readily to LD cycles that are more than a few hours shorter or longer than the period of the endogenous circadian rhythm. If the period of the LD cycle is too short or long for entrainment to occur, then the circadian rhythm will free-run with a period of the endogenous pacemaker.
One of the most widely used methods to examine how the LD cycle influences the circadian system has been to expose animals and humans maintained in constant conditions to pulses of light. The effects of the light pulse on a phase reference point of a circadian rhythm (e.g., onset of melatonin, minimum of body temperature) in subsequent cycles is then determined. The direction and magnitude of the phase shifts are strongly dependent on the circadian time at which the light pulse occurs. A phase–response curve (PRC) is a plot of the magnitude and direction of the time shift induced by an environmental perturbation as a function of the circadian time at which the perturbation is given. Light pulses presented near the onset of the subjective night (part of the circadian cycle that occurs during the dark or nighttime) delay circadian rhythms, whereas light pulses presented in the late subjective night or early subjective day (part of the circadian cycle that occurs during the light or daytime) advance circadian rhythms. Extensive studies have demonstrated that the LD cycle can entrain circadian rhythms, and that bright light can be used to manipulate human rhythms under a variety of experimental conditions.16,17 Although bright light (intensities approximating sunlight) is a very strong and reliable entraining agent for the circadian system,17 there is evidence that lower intensities, such as those encountered in ordinary room lighting can also affect the timing of human circadian rhythms.18 The discovery that in humans, both light-induced phase shifts and melatonin suppression are most sensitive to short wavelength light of approximately 460 nm19,20 provides an exciting new avenue for the development of light therapies to treat CRSDs.
Entrainment by Nonphotic Signals
The roles of activity and social cues as synchronizing agents for the human circadian system have been recognized since the early 1970s. Studies by Aschoff showed that scheduled bedtimes, mealtimes, and various timed social cues were able to entrain circadian rhythms.21 More recent studies indicate that sleep and social schedules may also phase shift the circadian clock.22 In addition, physical exercise during the night can produce a phase delay in human circadian rhythms.23 These findings indicate that scheduled social and physical activity programs may also be useful strategies for the treatment of CRSDs.
Melatonin
Melatonin is an important modulator of circadian rhythms,24 and it alters the timing of circadian rhythms in animals25 and humans.26 The circadian rhythm of melatonin production and release is controlled by the SCN via an indirect pathway, a noradrenergic synapse from the superior cervical ganglion to the pineal gland.27 The PRC for melatonin in humans indicates that administration of exogenous melatonin to humans in the early evening advances the phase of circadian rhythms, whereas administration in the early morning delays the phase, with the strongest phase-shifting effects of exogenous melatonin occurring during the evening, just preceding the increase in endogenous melatonin levels.28,29 In addition to its phase resetting properties, evidence supports a role for melatonin in sleep modulation by increasing evening sleep propensity and reducing core body temperature.24 Melatonin has been shown to decrease the firing rate of SCN neurons,30 and it has been proposed that by its inhibition of SCN firing, the increase of melatonin in the evening creates a sleep-permissive state.
The potential importance of melatonin for regulating the sleep–wake cycle has led to interest in its use for treatment of insomnia and CRSDs. Indeed, there is good evidence that melatonin may be effective for entraining circadian rhythms in blind people with free-running sleep–wake cycles31 and phase advancing circadian rhythms in people with DSPD.32 Another potential use for melatonin has been suggested from the role of the circadian system in maintaining consolidation of sleep during the early morning hours in the elderly.33
Delayed Sleep Phase Disorder, Delayed Sleep Phase Type, Delayed Sleep Phase Syndrome
Clinical Features
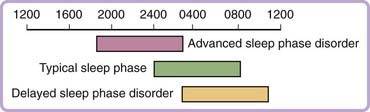
Delayed sleep phase disorder is characterized by sleep onset and wake times that are usually delayed 3 to 6 hours relative to conventional sleep–wake times (Fig. 41-1). The typical patient finds it difficult to initiate sleep before sometime between 2 and 6 AM and, when free of societal constraints, prefers wake times that are between 10 AM and 1 PM. Sleep itself is reported to be normal for the person’s age.34 These symptoms are chronic, usually of at least three months’—and quite often of many years’—duration. The clinical picture may be similar to sleep-onset insomnia. Patients are unable to advance their sleep times despite repeated attempts, and may report a history of prolonged sedative-hypnotic drug use, bedtime use of alcohol, behavioral interventions, or psychotherapy.35 Patients often report feeling most alert in the late evening and score highly as night people on a morningness-eveningness scale.36 Enforced “conventional” wake times may result in chronically insufficient sleep and excessive daytime sleepiness. Sleepiness is greatest in the morning and lessens as the circadian drive for wakefulness peaks in the late afternoon. In adolescents the syndrome may be associated with daytime irritability and poor school performance,37 whereas in adulthood, the syndrome may be associated with impaired job performance and associated financial difficulty, as well as with marital problems.38 DSPD may be mistaken for depression, in which the sleep–wake cycle may also be delayed (or advanced). Several studies, generally from psychiatric clinics, have emphasized an association of DSPD with mood and personality disorders.38,39
Epidemiology
DSPD has been reported as young as preadolescence and beyond sixty years of age.38 Although the actual prevalence of DSPD in the general population is not well characterized, evidence from one population based study indicates a prevalence of 0.17%.40 DSPD is reported to be more common in adolescents and young adults, with a reported prevalence of 7%,41 while in middle-aged adults, the prevalence may be one tenth of that, or 0.7%.42 In a sleep disorders clinic, 6.7%34 to 16%39 of patients seen for a primary complaint of insomnia were determined to have DSPD. There are no known gender differences in prevalence.
Pathogenesis
It has been pointed out that the tendency for late sleeping is not simply a function of the circadian drive for wakefulness interacting with the sleep homeostat, but rather is analogous to eating or other behaviors that are mandated by physiology but overlaid by varying individual emotional, social, and medical states.39 Although the exact cause of DSPD is not known, there have been several mechanisms proposed including both behavioral and physiological factors. Behavioral preference may play a major role in some cases of DSPD, particularly when bed times and rise times are not enforced.
In adolescence for example, the biological delay in the timing of circadian rhythms43 is likely exacerbated by late evening activities, such as doing homework, watching TV, and using the internet.44 Another factor may be the use of caffeine to combat sleepiness during normal waking hours. Staying up late and waking up late in the morning or early in the afternoon may result in an abnormal relationship between the endogenous circadian rhythm and the sleep homeostatic process that regulates sleep and wakefulness. Evidence also shows that under certain conditions (a background of dim light), ambient artificial light (as low as 100 lux) at night may be of sufficient intensity to affect circadian timing.18 Therefore, light exposure later in the evening may also perpetuate and exacerbate the phase delay. Furthermore, late wake times will delay exposure to light in the morning and may prevent active advancement of the circadian clock, allowing it to drift to a new phase relation with external clock time.
However, for many individuals with DSPD, symptoms often persist despite attempts to structure sleep and wake times, resulting in severe social or professional consequences,35 suggesting that behavioral factors alone do not fully explain this disorder. There is considerable evidence that DSPD is the result of alterations in the endogenous circadian system. For example, many physiologic markers of circadian phase persist in a delayed pattern despite enforced sleep–wake times,35 and there is also evidence that some individuals with DSPD have a hypersensitivity to nighttime suppression of melatonin by bright light.45 Reduced sensitivity of the oscillator to photic entrainment (i.e., a reduction in the amplitude of the advance portion of the PRC to light) has also been hypothesized, as has a prolonged free-running period length of the circadian cycle.35,46 Furthermore, the duration and timing of environmental light and dark exposure may play a role in the expression of the DSPD phenotype. For instance, the prevalence of DSPD may be increased at extreme latitudes.47 DSPD has also been reported following minor traumatic brain injury.48 There is also evidence for a genetic basis to DSPD. In some cases the syndrome may be familial presenting with an autosomal dominant mode of inheritance.49 Further support for a genetic basis for DSPD derives from reports of polymorphisms in circadian genes such as, hPer3, arylalkylamine N-acetyltransferase gene, HLA genes, and Clock.50–53
Although it is commonly accepted that DSPD is predominantly a result of alterations of circadian timing, there is recent evidence that alterations in the homeostatic regulation of sleep may play an important role as well.54,55 Polysomnographic recordings of sleep in individuals with DSPD have shown that sleep architecture is not disrupted after the initiation of sleep when subjects are allowed to sleep until their desired wake times.37,56 However, following 24 hours of sleep deprivation, subjects with DSPD, when compared to controls, show a decreased ability to compensate for sleep loss during the subjective day and the first hours of subjective night.54,57 Therefore, it is likely that both alterations in circadian timing and impaired sleep recovery contribute to symptoms of insomnia and excessive sleepiness in DSPD.
Diagnosis
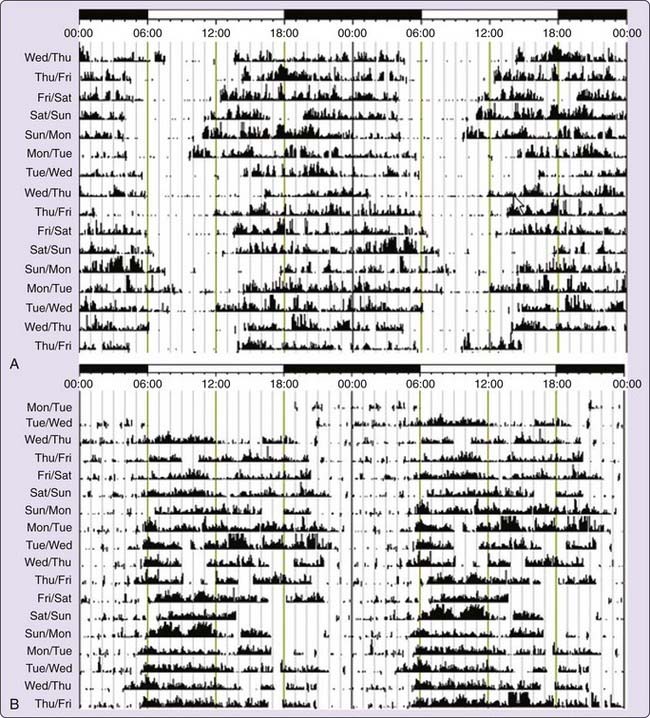
The diagnosis of DSPD is usually made on the basis of the patient’s history of chronic or recurrent complaint of symptoms of insomnia due to a stable delay in the timing of the major sleep and wake period.58 The sleep disturbance is associated with impairment of social, occupational, or other areas of functioning. In addition, sleep log or actigraphy monitoring should be performed for at least 7 days to demonstrate a stable delay in the timing of the habitual sleep period. Actigraphy is a practical tool for assessing sleep–wake cycles relative to clock time, and has become more widely available clinically (Fig. 41-2A).59 A morningness-eveningness questionnaire, such as the Horne-Ostberg questionnaire36 or the Munich chronotype60 questionnaire, may be useful in confirming the patient’s circadian preference, but is not required to make the diagnosis.61,62
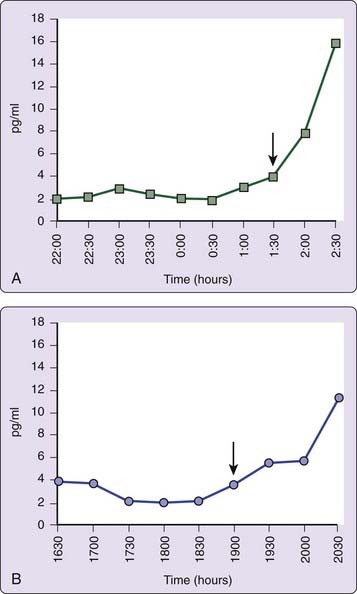
The use of other physiologic markers of circadian timing such as continuous recording of body temperature,63 or dim light plasma melatonin onset (DLMO)64 may also aid in determining the phase relation of circadian and terrestrial time, although routine clinical availability remains limited. DLMO is probably the most useful marker for circadian pacemaker output.65 Individuals with DSPD usually have DLMO times that occur after 10 PM (Fig. 41-3A).64,66,67 Determination of DLMO can be made by measurements of melatonin from plasma or saliva. Commercially available salivary determination of DLMO for clinical use may be feasible in the near future.
Treatment
The goal of therapy is to align the timing of the circadian clock with the desired 24-hour LD cycle. Richardson6 has noted that treatment of DSPD is the same whether it is the result of a primary behavioral or a primary physiologic process. Adherence to good sleep hygiene principles and identification and treatment of comorbid medical and psychiatric disorders are essential components in the management of DSPD. In addition, chronotherapy, light therapy, and pharmacological agents, such as melatonin, have been shown to be useful treatments for patients with DSPD.
Chronotherapy
The use of chronotherapy has been successful in a small group of patients in a laboratory setting.35 Chronotherapy requires a successive delay of sleep times by 3 hours daily over a 5 to 6 day period until the desired sleep time is achieved. This shift is followed by rigid adherence to a set sleep–wake schedule and good sleep hygiene practices. However, outside the laboratory setting, the potential confounding effects of light exposure at the wrong circadian time may limit the effectiveness and practicality of this approach.6
Light
As described earlier, light plays a major role in resetting the human circadian pacemaker.16,17,68 Therapy with bright light in the morning (the advance portion of the human PRC) should advance the phase of circadian rhythms in DSPD, and may be more practical than chronotherapy.69 Although a number of reports of successful application of bright light therapy in DSPD exist,70–73 large, randomized, placebo-controlled studies to determine the intensity, duration, and overall effectiveness are still needed. Rosenthal and colleagues found that 2 hours of bright light exposure (2500 lux) in the morning (7 to 9 AM), together with light restriction in the evening, successfully phase advanced (by 1.4 hours) circadian rhythms of core body temperature and multiple sleep latencies in 20 patients chosen prospectively after meeting clinical criteria for DSPD.73 In contrast, a retrospective report from a referral sleep clinic found that only 7 of 20 patients with DSPD treated with bright light alone were able to entrain reliably to a desired sleep schedule.
The human PRC to a single 3-hour bright light pulse suggests that a light pulse given slightly before the time of body temperature minimum will result in a maximal phase delay, whereas a pulse given slightly after the minimum will cause a maximal phase advance (each about 2 hours).68 When light pulses over three successive cycles were used, larger shifts (4 to 7 hours) can be produced. Because body temperature minimum is not routinely measured clinically, light therapy is usually timed using sleep logs to estimate the patient’s endogenous circadian phase. A light pulse of 1 to 2 hours’ duration and between 2500 and 10,000 lux is usually administered toward the end of the sleep–wake cycle. Because the portion of the PRC at which the greatest phase advance can be achieved occurs during sleeping hours, light is usually given immediately upon awakening in the morning, which results in a smaller phase advance. However, in severely delayed individuals the sleep–wake cycle may not necessarily correlate with circadian phase; therefore, early morning light could in theory be inadvertently given on the delay portion of the PRC, worsening the problem. Regenstein and Pavlova39 reported a patient who slept later after receiving light exposure at 6 AM. Another factor that may limit the practicality of bright light treatment is that many individuals with DSPD may find it difficult to wake in time for administration of bright light therapy.39
Although the timing, intensity, and duration of light exposure have not been fully established in clinical practice, there is sufficient evidence to support the efficacy of bright light therapy for the treatment of DSPD. The recent practice parameters established by the American Academy of Sleep Medicine considered light therapy as a treatment guideline for DSPD.61

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


