Figure 21.1. Graphic representation of continuous spike-and-wave during sleep evolution over time. The age of clinical events varies in individual patients but tends to peak at approximately the displayed ages. EEG, electroencephalogram; R, regression; SF, seizure freedom; SO, seizure onset; y, years.) (Modified from Sánchez Fernández I, Loddenkemper T, Peters JM, et al. Electrical status epilepticus in sleep: clinical presentation and pathophysiology. Pediatr Neurol. 2012;47(6):390–410.)
Seizures are the most common presenting sign in CSWS. The age at seizure onset is typically between 2 and 4 years with a bimodal distribution. Seizure onset clusters at around 2 years of age in patients with underlying structural brain lesions and at around 4 years of age in patients with CSWS of unknown etiology (2). During the prodromal stage, seizures are infrequent and relatively easy to control, and development is not severely affected. Seizures typically occur years before the ESES pattern is recognized on EEG. Therefore, recognition of CSWS during the prodromal stage does not typically occur.
Subacute worsening involving seizures, developmental regression, and worsening of EEG abnormalities occurs at approximately 5 to 6 years of age. This subacute regression heralds the acute stage. During the acute stage, seizures become more frequent and difficult to control, patients suffer a global and severe developmental regression, and almost continuous spiking during non-REM sleep appears on the EEG. CSWS is typically diagnosed during this acute stage. After a variable number of years, seizures tend to resolve spontaneously with a peak time of seizure freedom at approximately 6 to 9 years of age (2).
In the residual stage that follows seizure freedom, there is a gradual normalization of the EEG pattern and mild-to-moderate cognitive improvement, although severe residual cognitive impairments often remain (2,4).
Seizures.
During the prodromal stage, seizures typically occur out of sleep (3). Seizures are infrequent and most patients have only one seizure type. Seizure types during this prodromal stage include focal or unilateral motor seizures, focal dyscognitive seizures, absence seizures, and generalized tonic–clonic seizures. During the acute stage, most patients manifest several seizures of several different types per day, and seizures are often difficult to control (2,4,14). Unilateral seizures become rare, atonic and bilateral motor seizures appear, and atypical absence seizures increase in frequency. Absence seizures may also evolve into absence status epilepticus (2,4). The lack of tonic seizures is a typical feature of CSWS and permits differentiation from patients with Lennox–Gastaut syndrome (2,4).
Development.
Developmental regression in CSWS involves a wide spectrum of behavioral, cognitive, language, social, and motor milestones in varying but often severe degrees. Regression includes a decrease in overall intelligence, learning disorders, memory problems, and attention span (1,4,15,16). Language regresses in the form of a subacute and progressive aphasic disorder with spontaneous fluctuations over time. Behavior becomes disruptive, and behavioral manifestations include hyperactivity, impulsivity, and even aggression. Deterioration of fine and gross motor skills as well as loss of social skills add to the severe functional deterioration.
EEG Characteristics
Evolution of the EEG Characteristics.
The EEG hallmark of CSWS is ESES, a pattern of almost continuous spike-and-wave that occupies most or all of the EEG tracing during non-REM sleep (2–4). However, the ESES pattern is typically preceded by earlier and milder EEG abnormalities. In the few patients who underwent an EEG during the dormant stage, mild EEG abnormalities and infrequent epileptiform discharges have been detected. During the prodromal stage, epileptiform discharges are rare and occur essentially only during sleep. This pattern worsens progressively, and epileptiform discharges become also abundant during wakefulness, with additional marked potentiation of epileptiform discharges in the transition from wakefulness to sleep. A clinical presentation with almost continuous spike-and-wave during non-REM sleep is typical during the acute stage (Fig. 21.2).
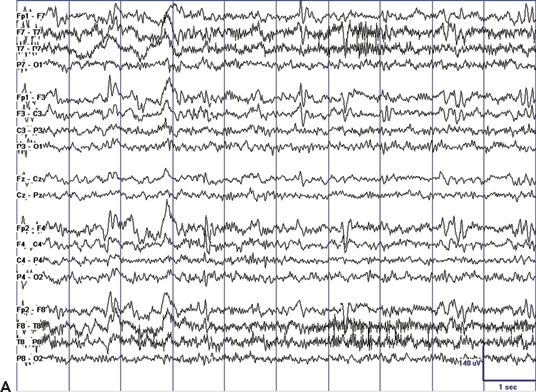
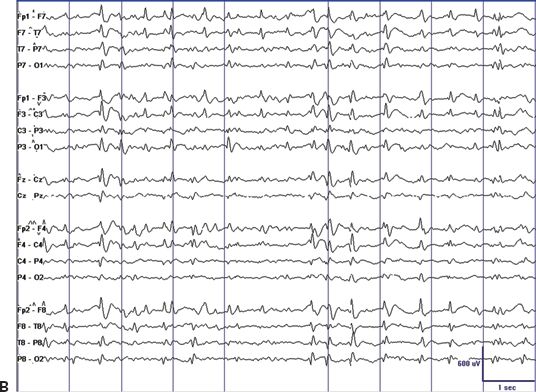
Figure 21.2. EEG tracings illustrating the dramatic sleep potentiation of epileptiform activity in the transition from wakefulness to sleep. A: During wakefulness, infrequent and relatively low-amplitude spikes occur in the setting of a normal background. B: During non-REM sleep, almost continuous spike-and-wave replace the normal sleep graphoelements of non-REM sleep. Also note the difference in voltage.
Classical Definition of the ESES Pattern.
The EEG pattern of ESES is classically defined by the presence of generalized bilateral and symmetric 1.5- to 3-Hz spike–waves that occupy at least 85% of the EEG tracing during non-REM sleep (4,17). The originally proposed cutoff value of 85% has largely been followed in the literature. However, the definition of the International League Against Epilepsy does not include a specific threshold percentage and only requires that spike-and-wave be “continuous” and “diffuse” (18). Consequently, there have been authors who used lower cutoff values (6,7,19,20) or did not consider a specific threshold (14). Further, the percentages of epileptiform activity vary over time in the same patient (13). Therefore, a more flexible criterion such as “a dramatic activation of epileptiform discharges by sleep” may be considered in the definition of ESES in the future (1).
Quantification of Epileptiform Activity.
The classical quantification of epileptiform activity in ESES is the spike–wave index, expressed as a percentage of slow sleep occupied by spike–waves (3,4). Most authors refer to this percentage without defining the method or the EEG segment considered for calculation. Quantification of epileptiform activity in ESES can be assessed as the percentage of 1-second bins with at least one spike-and-wave in each second (13,21) or as the total number of spike–waves per unit of time (13). Another difficulty in the reproducibility of methods for quantification of epileptiform activity is that there is no consensus regarding the specific portion of the EEG tracing used for the calculation of epileptiform activity: Some authors use whole-night sleep recordings, and others use selected segments of non-REM sleep (5). Different timings of EEG assessment with respect to sleep and circadian phases as well as different methods of quantification may result in considerable variation in percentages among studies. Efforts are under way to create a better defined and reproducible method of epileptiform activity quantification.
Lateralization of Epileptiform Activity.
The first descriptions of the ESES EEG pattern required bilateral, symmetric, or diffuse EEG manifestations (17,18). However, markedly asymmetric, bilateral, or even more focal examples of ESES have been published, and series that compared the clinical features of patients with generalized versus those with more focal ESES did not find marked differences in clinical presentation (7,14,22).
Criteria for ESES.
Based on the above considerations, three essential features emerge when describing the EEG pattern of ESES: (i) a marked activation or potentiation of epileptiform discharges in the transition from wakefulness to sleep, (ii) leading to a (near)-continuous pattern of slow spike-and-wave, and (iii) that occupy a “significant proportion” of the non-REM sleep EEG tracing with a cutoff value ranging from 25% to 85% in the literature (2).
Age of ESES in the EEG.
The age at detection of the ESES pattern depends both on the definition of this pattern as well as on the frequency of routine, prolonged, or overnight EEG recordings with sufficient periods of sleep captured in the EEG tracing. With these limitations in mind, the ESES pattern appears at approximately 4 to 8 years of age and remits at around 8 to 9 years of age, broadly coinciding in time with the acute stage of CSWS (2).
Pathophysiology and Etiology
The cause of CSWS and the pathophysiologic mechanisms that lead to age-specific clinical and EEG abnormalities are unknown. The ESES pattern is thought to be related to a disruption of the physiologic corticothalamocortical circuitry, which may also be involved in the generation of sleep spindles (23). When structural or functional insults disrupt this circuitry, physiologic sleep spindles may be replaced by generalized spike-and-wave, and, in some cases, evolve to ESES.
It is currently unknown to what extent this EEG pattern is causal in developmental regression seen in CSWS and whether effective treatment of ESES improves developmental prognosis in patients with CSWS. Early developmental structural brain lesions have been identified as one important etiology in CSWS, with genetic predisposition possibly playing an additional role.
Early Developmental Lesions
Several case reports and small case series have found an association between CSWS and malformations of cortical development (24) and with early vascular lesions that involve the thalamus and basal ganglia (25–27). Larger studies also support the hypothesis that insults to the developing brain are associated with the development of the ESES pattern on EEG and with CSWS. In a series that studied 32 patients with prenatal or perinatal thalamic lesions, a dramatic sleep potentiation of epileptiform activity occurred in 29 cases (90.6%) (28). In a series of 67 patients with ESES and neuroimaging, 33 patients (49.3%) presented with early developmental lesions, with no information on thalamic involvement (7). In another series, 18 of 44 (41%) patients with ESES had structural lesions of the brain, but no details on thalamic involvement were available (29). In a series of 14 survivors of neonatal thalamic hemorrhage secondary to cerebral sinovenous thrombosis, 7 (50%) had sleep potentiation of epileptiform activity (30). In another series of 100 patients with ESES, 48 (48%) had early developmental lesions and 14 of these demonstrated thalamic involvement (31). In this series, patients with ESES were compared with a control group of patients with epilepsy but without ESES (31). Patients with ESES had a higher frequency of early developmental lesions (48% vs. 19%, odds ratio = 3.9, 95% CI: 1.71 to 8.9) and a higher frequency of thalamic lesions (14% vs. 2%, odds ratio = 7.49, 95% CI: 0.95 to 58.76) than did patients with epilepsy without ESES (31). In all of the above-mentioned series, the lesion occurred typically perinatally or early in life, with a predominance of vascular etiology in all series (Fig. 21.3). Taken together, these studies demonstrate that vascular brain lesions affecting the thalamus early during brain development are common in patients with ESES and may potentially play a causal role. However, these series also indicate that in more than half of patients, the cause cannot be attributed to a macroscopic structural brain abnormality. In those cases, a similar functional disruption of the corticothalamocortical neuronal networks without macroscopic correlate on MRI may play a role.
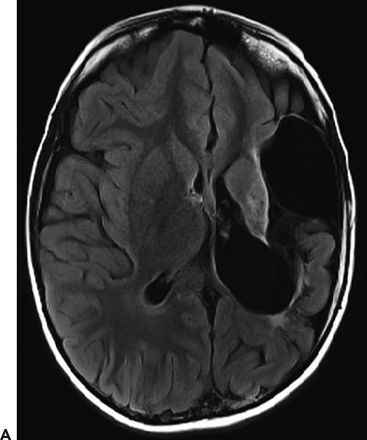
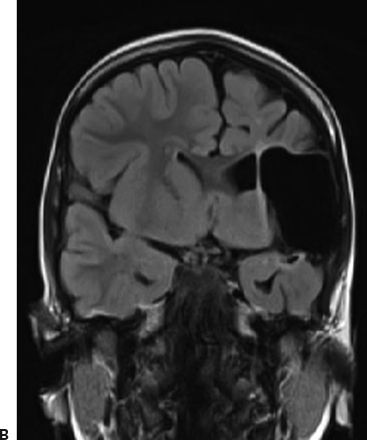
Figure 21.3. Early developmental lesion in a patient with CSWS. An extensive cystic encephalomalacia affects the left hemisphere in the distribution of the left middle cerebral artery. The patient suffered a perinatal stroke. A: Axial view, T2-weighted FLAIR MRI. B: Coronal view, T2-weighted FLAIR MRI.
Evolution into CSWS from Other Epileptic Syndromes
“Benign” pediatric focal epilepsy syndromes (“benign” epilepsy of childhood with centrotemporal spikes, Panayiotopoulos syndrome, and Gastaut-type late-onset childhood occipital epilepsy) share some common features with CSWS and LKS:
1. Age-related evolution
2. Epileptiform discharges that are of disproportionate severity in comparison with the associated clinical features
3. Sleep potentiation of epileptiform activity
4. Persistence of epileptiform discharges for years after seizure freedom.
5. Because of these similarities, it has been proposed that “benign” pediatric focal epileptic syndromes may represent a milder variation within this electroclinical spectrum.
A shared pathophysiology with different severities could potentially account for the electroclinical continuum of CSWS, LKS, and “benign” focal pediatric epilepsy syndromes. Furthermore, a genetically determined disruption of brain maturation during the first years of life may lead to hyperexcitable neuronal networks in selected cases, while other insults may also play a role. The location of the disrupted and hence hyperexcitable network then translates into different clinical features: the lower rolandic cortex that represents the face and the oropharynx could lead to the clinical features of “benign” epilepsy with centrotemporal spikes, the central autonomic neuronal networks could manifest their discharges as Panayiotopoulos syndrome, the occipital lobe that represents the cortical visual system could express their abnormalities as Gastaut-type late-onset childhood occipital epilepsy, and the involvement of the language cortex could manifest as LKS (32,33). More severe disruptions of neuronal networks as seen due to larger early developmental lesions could lead to CSWS (23,28,31). This framework of a common “seizure susceptibility syndrome” suggests that most affected children (>90%) manifest only with epileptiform discharges without clinical correlate, around 9% may present with benign pediatric focal epileptic syndromes, and in <1% of children, frequent and difficult-to-control seizures and severe neuropsychological regression may occur (33).
Frequently, patients present with electroclinical characteristics that are intermediate between the different syndromes. Further, patients with CSWS often initially present with “benign” pediatric focal epilepsy syndromes or other forms of epilepsy that later evolve into CSWS (14,34).
Genetic Predisposition
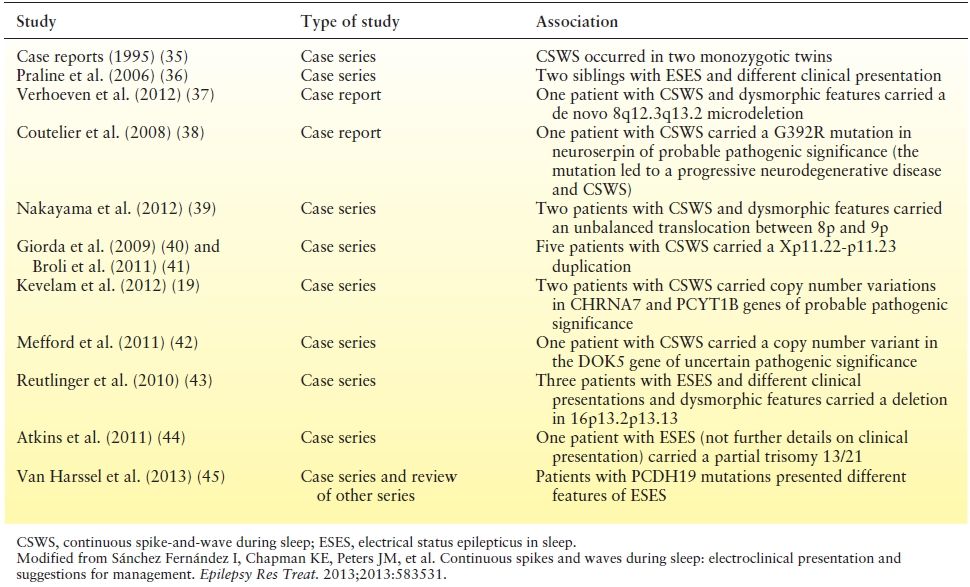
Genetic factors have been implicated in selected cases of CSWS and familial antecedents of seizures (including febrile seizures) are found in approximately 10% to 15% of the cases (4,32). A growing number of case reports and small series describe associations with copy number variations and different mutations in several chromosomes (Table 21.1). To date, the causal role of these heterogeneous genetic factors in CSWS is largely undefined. Some of these genetic variants may not be associated with CSWS per se, but with different neurologic conditions that result in the final common pathway of ESES and/or CSWS. Recently, heterozygous mutations in GRIN2A (codifying for the NMDA receptor) have been found in 27 of 359 (7.5%) patients with “seizure susceptibility syndrome” and appear to be specifically correlated with its pathophysiology (33).
Table 21.1 Genetic Factors That Have Been Described in Association with CSWS or with ESES
Treatment
Treatment for CSWS is based on case reports and small uncontrolled series. Comparison of treatment strategies between different series is challenging because of heterogeneous patient populations, different drug doses, frequent polytherapy, variable durations of treatment, different measures of outcome, and naturally occurring fluctuations in severity over time. We will review the characteristics of different treatment options and then provide general suggestions for management.
Benzodiazepines
Benzodiazepines have demonstrated efficacy in reducing epileptiform activity. Transitory resolution of the ESES pattern was observed after the administration of clonazepam (6,8). However, the shorter half-life of diazepam may have contributed to a more frequent use in CSWS. In a series of four patients with CSWS refractory to valproate and ethosuximide, a short cycle of high-dose oral or rectal diazepam (0.5 to 1 mg/kg/day for 6 to 7 days) showed short-term efficacy in two patients (6). In a series of 15 patients with CSWS, all patients responded to high-dose rectal diazepam (46). High-dose oral diazepam (0.75 to 1 mg/kg/day for 3 weeks) was also efficacious in 3 out of 8 (37.5%) patients, but the response was temporary (14). In 29 patients with ESES and different clinical presentations, the mean epileptiform activity decreased from 77% to 41% after nocturnal administration of 1 mg/kg of oral diazepam (47). This reduction in epileptiform activity persisted for several months (48), but whether this reduction in epileptiform activity is accompanied by a sustained improvement in clinical neuropsychological features remains to be proven. Other series showed that 9 of 10 patients did not respond to valproate and benzodiazepines, and 3 patients experienced adverse behavioral side effects (16). Adverse effects of high-dose diazepam treatment are generally considered mild and self-limited (46,47), but severe behavioral disinhibition that at times necessitates discontinuation have also been described in a few children (1,48). Small series suggest that the benzodiazepine derivative clobazam is also successful in the treatment of CSWS (30) and deserves further study.
Conventional Antiepileptic Drugs
The most commonly used antiepileptic drugs for CSWS include valproate, ethosuximide, and levetiracetam (49). In a series of 15 patients with CSWS treated with high-dose valproate alone or with valproate and ethosuximide, 10 cases (67%) responded with long-term control of their epilepsy and partial recovery of cognitive function (6). In a separate study, the combination of valproate and ethosuximide was effective in two additional patients (50). In contrast, other series did not report similar improvements after treatment with comparable medication regimens. Valproate was not effective in a series of 28 patients (14). Valproate and benzodiazepines did not lead to any improvement in seven patients and were associated with adverse behavioral reactions in three children (16). Levetiracetam was effective in several case series and also in the only placebo-controlled double-blind crossover study in patients with ESES. In this cross-over study of 18 patients, levetiracetam reduced epileptiform activity from a spike index of 56 to 37, although 3 other patients discontinued treatment because of negative cognitive side effects (51). Other drugs with some effectiveness in small series include sulthiame (14,52) and lamotrigine (24,53). Most clinicians avoid phenytoin, phenobarbital, and, especially, carbamazepine and oxcarbazepine because these drugs have been associated with exacerbation of epileptiform discharges in patients with ESES (53,54). In particular, carbamazepine and oxcarbazepine have been associated in some patients with “benign” focal epilepsies of childhood with deterioration and transformation into CSWS, although a clear causal effect has not been demonstrated (7,55).
Corticosteroids
Corticosteroids lead to improvement or even resolution of ESES in selected cases of CSWS. In a series of 44 children with ESES and clinical presentations of variable severity, prolonged corticosteroid treatment (hydrocortisone 5 mg/kg/day during the first month, 4 mg/kg/day during the second month, 3 mg/kg/day during the third month, and 2 mg/kg/day during the next 9 months followed by slow withdrawal for a total treatment duration of 21 months) led to reductions of seizures or neuropsychological improvement in 34 of 44 (77.3%) cases, with 34 patients achieving complete control of seizures and normalization of EEG abnormalities in 21 patients. The long-term remission rate was 45% (29). Inclusion of milder clinical presentations in this series may make it difficult to compare the results of this study with the results of other CSWS series that included patients with more prominent global cognitive deterioration (29). In a different study, 11 of 17 patients with CSWS responded to different corticosteroids (prednisone, methylprednisolone, or adrenocorticotropic hormone) (14). The intramuscular administration of 0.001 to 0.04 mg/kg/day of adrenocorticotropic hormone was reported to be effective in one of four patients (6). Although generally regarded effective, the side effects of corticosteroids frequently limit their long-term use.
Immune Modulation Therapy
Very limited data are available on intravenous immunoglobulins, but some series report a response to treatment in a few cases. Variable outcome results, high cost, and risks of complications associated with immunoglobulins may have prevented widespread use in the treatment of CSWS to date (14,56).
Surgery
Selected patients with CSWS may benefit from surgical treatment. In CSWS, the ESES pattern and the seizures tend to spontaneously resolve with time, and therefore, ESES as a sole indication for epilepsy surgery is debated (24). However, in patients with severe developmental regression, a well- defined structural lesion and ongoing intractable seizures, epilepsy surgery may be a very effective treatment for seizures and epileptiform activity, with some improvement in developmental regression (11). Potential surgical interventions include multiple subpial transections (MSTs), focal resective surgery of the epileptogenic zone, hemispherectomy, and corpus callosotomy. MST consists of multiple small superficial parallel cuts in the cortex that theoretically sever only the local corticocortical connections in an attempt to disrupt local horizontal epileptic circuitry without altering the vertical neural columns and their function. MSTs led to recovery of age- appropriate speech in 7 of 14 patients with ESES and acquired aphasia (57) whereas other series report less prominent language improvement (58,59). Two patients with CSWS secondary to neonatal stroke markedly improved after hemispherectomy (60). In another study, two patients with CSWS secondary to early developmental lesions in the thalamus became seizure free after a hemispherectomy in one and after an extensive corticectomy around a large porencephalic cyst in the other (28). Additionally, 13 patients with CSWS secondary to different early developmental lesions underwent various surgical procedures including anterior callosotomy (6 patients), complete callosotomy (3 patients), hemispherectomy (2 patients), and lobar resection (2 patients). Subjects achieved an overall improvement in seizure control and EEG features in most cases (15). Improvements after surgery included resolution of ESES, seizure freedom or improved seizure control, and improvements in cognitive function, with varying results among patients. Data on the long-term neurocognitive outcome of surgically managed CSWS patients are not available.
Suggestions for Management
The current literature does not permit the development of an evidence-based management approach to CSWS (Fig. 21.4). In practice, most patients with CSWS were already on a standard antiepileptic drug (valproate, levetiracetam, etc.) after seizure onset and prior to the diagnosis of CSWS. Once patients enter the acute phase, standard antiepileptic drugs, corticosteroids, and benzodiazepines may be considered as first choices depending on the particular patient and the familiarity of the physician with these drugs. Several groups have reported the usefulness of benzodiazepines, and one protocol includes application of nocturnal diazepam 1 mg/kg up to 40 mg during the first night while monitoring vital signs in the hospital followed by 0.5 mg/kg (maximum: 20 mg) upon discharge and every following night for 1 to 3 months (47,48). For chronic CSWS management, and particularly for seizure control, standard antiepileptic drugs such as valproate, levetiracetam, and ethosuximide are frequently used. Polytherapy is often needed. Medication selection should be guided by presenting seizure types. Other options include treatment with corticosteroids or adrenocorticotropic hormone. Epilepsy surgery should be considered, especially in patients with an early unilateral developmental lesion, even when the epileptiform activity on EEG is generalized. For the acute control of very active nighttime epileptiform discharges, high-dose benzodiazepines have been used over a period of a few months. While adequate control of seizures improves the quality of life of the patients and should be pursued, it is unknown how aggressively interictal epileptiform activity in relationship with neurocognitive regression should be treated. Only prospective studies that correlate the response to treatment of interictal epileptiform activity with the improvement in neurocognitive function will be able to answer that relevant question.
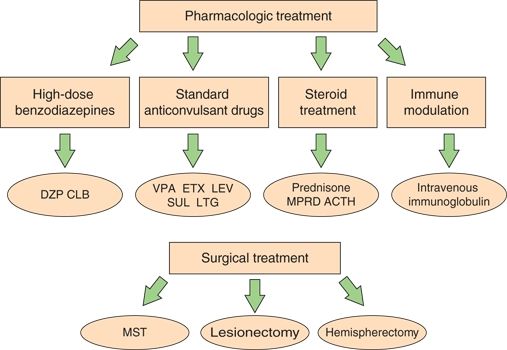
Figure 21.4. Options for the management of patients with CSWS. Options for chronic management are high-dose benzodiazepines, standard antiepileptic drugs in different combinations, and corticosteroids and immune-modulating agents. These options are considered first choice by different authors, although standard antiepileptic drugs are generally started prior to diagnosis of CSWS. Epilepsy surgery is reserved for a few selected refractory cases. ACTH, adrenocorticotropic hormone. (From Sánchez Fernández I, Chapman KE, Peters JM, et al. Continuous spikes and waves during sleep: electroclinical presentation and suggestions for management. Epilepsy Res Treat. 2013;2013:583531.)
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


