Neurodegenerative Proteinopathies
Progressive Supranuclear Palsy
Creutzfeldt-Jakob (Prion) Disease
Chronic Traumatic Encephalopathy
Bilateral Posterior Cerebral Artery Occlusion
Dissociative (Psychogenic) Amnesia
Alcoholic Korsakoff Amnestic Syndrome
Paraneoplastic and Cell-Surface-Directed Antibody-Mediated Limbic Encephalitis
Dementia is an acquired, generalized, and usually progressive impairment of cognitive function. Dementia differs from other disorders of cognitive function, such as coma (see Chapter 3, Coma) or confusional states (see Chapter 4, Confusional States), in that the level of consciousness (wakefulness or arousal) is preserved in dementia. Although the prevalence of dementia increases with advancing age (Figure 5-1), dementia is not an invariable consequence of aging and results instead from diseases involving the cerebral cortex, its subcortical connections, or both. Normal aging may be associated with minor alterations in neurologic function (Table 5-1) and with neuroanatomic changes, such as enlargement of cerebral ventricles and cortical sulci seen on computed tomography (CT) or magnetic resonance imaging (MRI) scans (Figure 5-2). However, these are not indicative of dementia. The term mild cognitive impairment (MCI) is sometimes used to describe deficits that are more severe than are customarily seen with normal aging but are insufficiently pronounced to warrant a diagnosis of dementia. Nevertheless, patients with MCI have an increased risk (approximately 10% per year) of developing dementia.
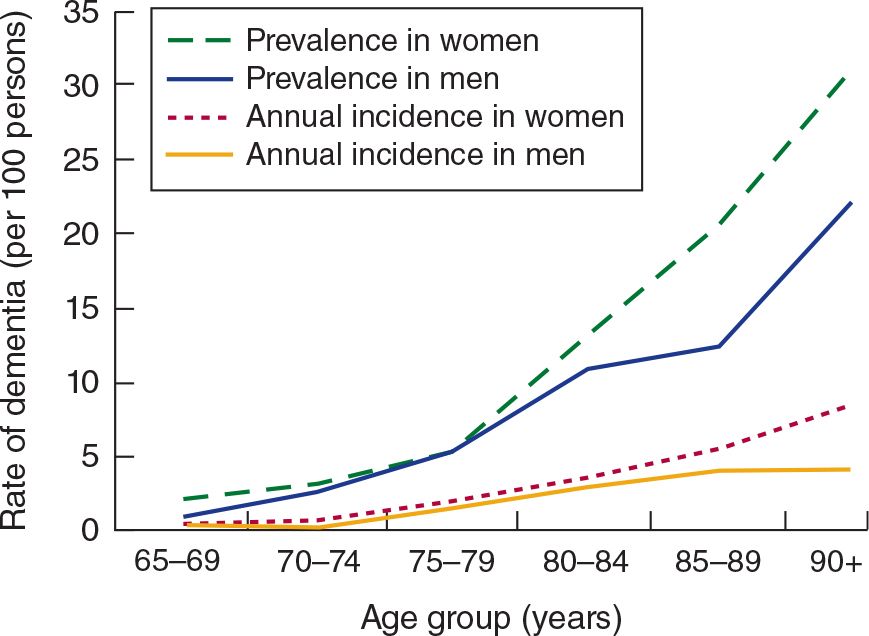
![]() Figure 5-1. Relationship between advancing age and incidence and prevalence of dementia. (Used with permission from Halter JB, Ouslander JG, Tinetti ME. Hazzard’s Geriatric Medicine and Gerontology. 6th ed. New York, NY: McGraw-Hill; 2009.)
Figure 5-1. Relationship between advancing age and incidence and prevalence of dementia. (Used with permission from Halter JB, Ouslander JG, Tinetti ME. Hazzard’s Geriatric Medicine and Gerontology. 6th ed. New York, NY: McGraw-Hill; 2009.)
Table 5-1. Neurologic Changes in Normal Aging.
Cognitive
Slowed information processing
Impaired learning and recall of new information
Reduced spontaneous word finding and verbal fluency
Increased reaction time
Neuro-ophthalmologic
Small, sluggishly reactive pupils
Impaired upgaze
Impaired convergence
Motor
Atrophy of intrinsic hand and foot muscles
Increased muscle tone
Flexion (stooped) posture
Small-stepped or broad-based gait
Sensory
Reduced visual acuity
Reduced auditory acuity
Reduced gustatory acuity
Reduced olfactory acuity
Reduced vibration sense
Reflexes
Primitive reflexes
Absent abdominal reflexes
Absent ankle reflexes
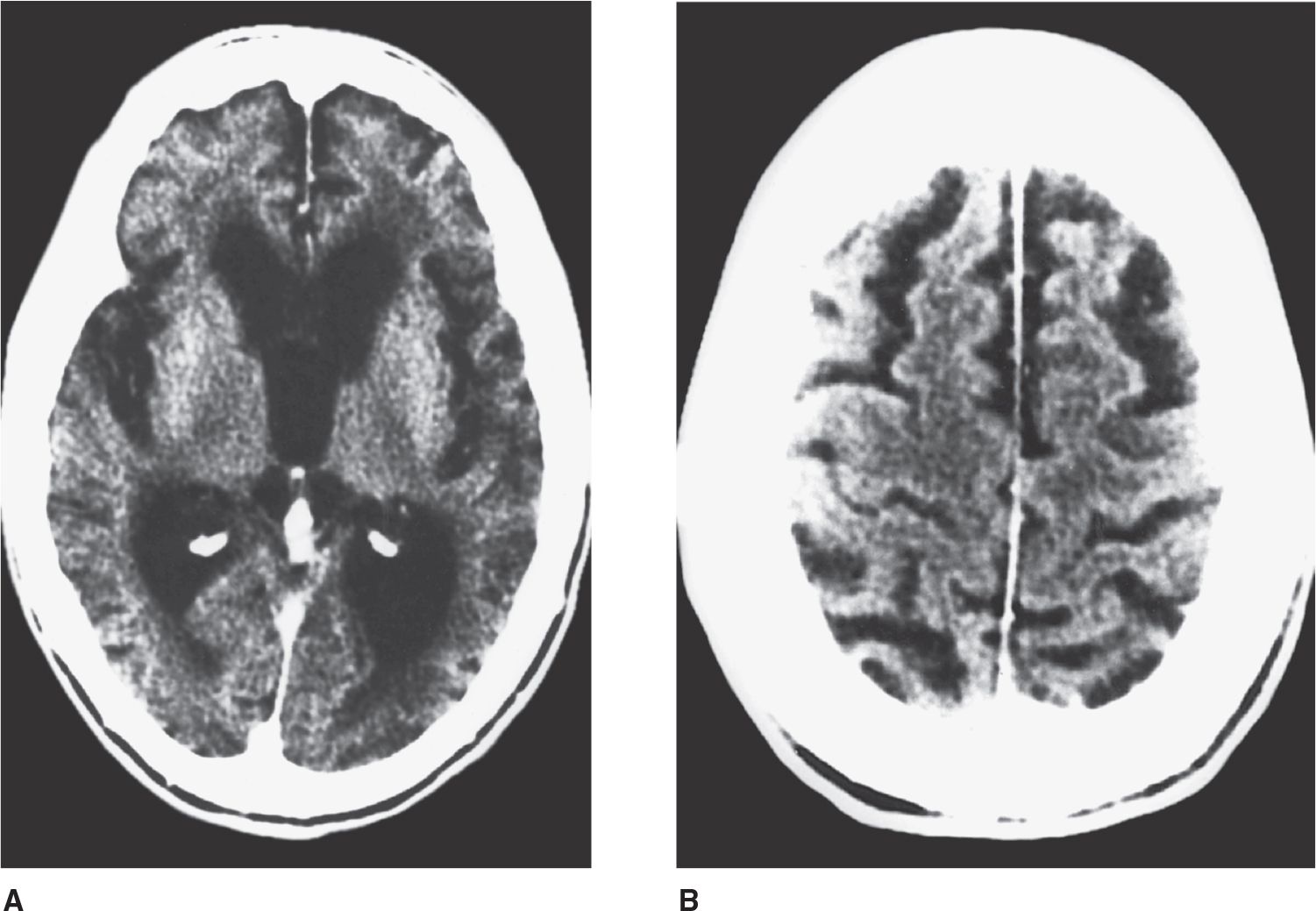
![]() Figure 5-2. CT scan in cerebrocortical atrophy, showing ventricular dilation (A) and prominent cortical sulci (B).
Figure 5-2. CT scan in cerebrocortical atrophy, showing ventricular dilation (A) and prominent cortical sulci (B).
In contrast to dementia, which affects multiple spheres of cognitive function, more limited disorders of cognition may also occur. These include deficits in language function (aphasia) or motor (apraxia) or sensory integration, which are considered in Chapter 1, Neurologic History & Examination. Memory disturbance (amnestic disorder or amnesia), another example of a circumscribed cognitive defect, is discussed in this chapter. Memory may also be impaired in normal aging and in dementia, but in the former impairment is mild, and in the latter it is accompanied by defects in, for example, reasoning, judgment, behavior, or language. Some causes of dementia, notably Alzheimer disease, produce early and disproportionate impairment of memory and, at least in the early stages of disease, may be difficult to distinguish from a pure amnestic disorder.
APPROACH TO DIAGNOSIS
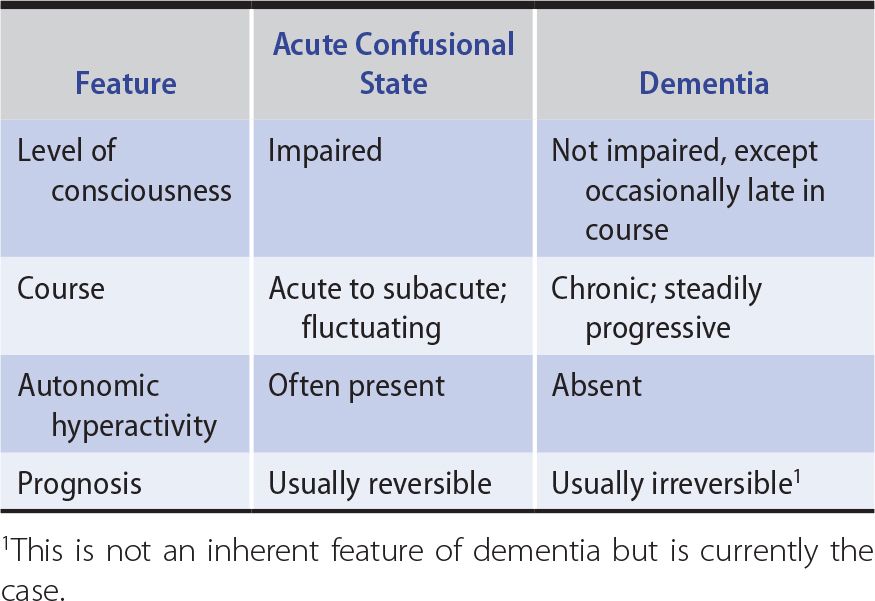
The first step in evaluating a patient with cognitive impairment of any kind is to determine the nature of the problem, which should be classified as affecting the level of consciousness (confusional state or coma) or the content of consciousness. Table 5-2 lists key differences that may be useful in making this distinction. If the content of consciousness is impaired, a global cognitive disorder (dementia) should then be distinguished from a more circumscribed deficit, such as amnesia or aphasia. This distinction is important because classification of the disorder determines the diagnostic approach.
Table 5-2. Differences Between Acute Confusional States and Dementia.
In some cases, it may be difficult to distinguish dementia from a psychiatric disturbance (pseudodementia). Pseudodementia due to psychiatric disease is discussed later in this chapter.
The final step in the diagnosis of dementia or an amnestic syndrome is to identify the specific cause. Although the greatest emphasis should be on finding a treatable cause, even untreatable causes can be important to identify. At present, only approximately 10% of dementias are reversible, but the extent to which the quality and duration of life can be improved in these cases justifies the effort and expense required to detect them. Diagnosis may be important in untreatable disorders as well, to provide the patient and family with prognostic information or genetic counseling, or to alert family members and medical personnel to the risk of a transmissible disease.
HISTORY
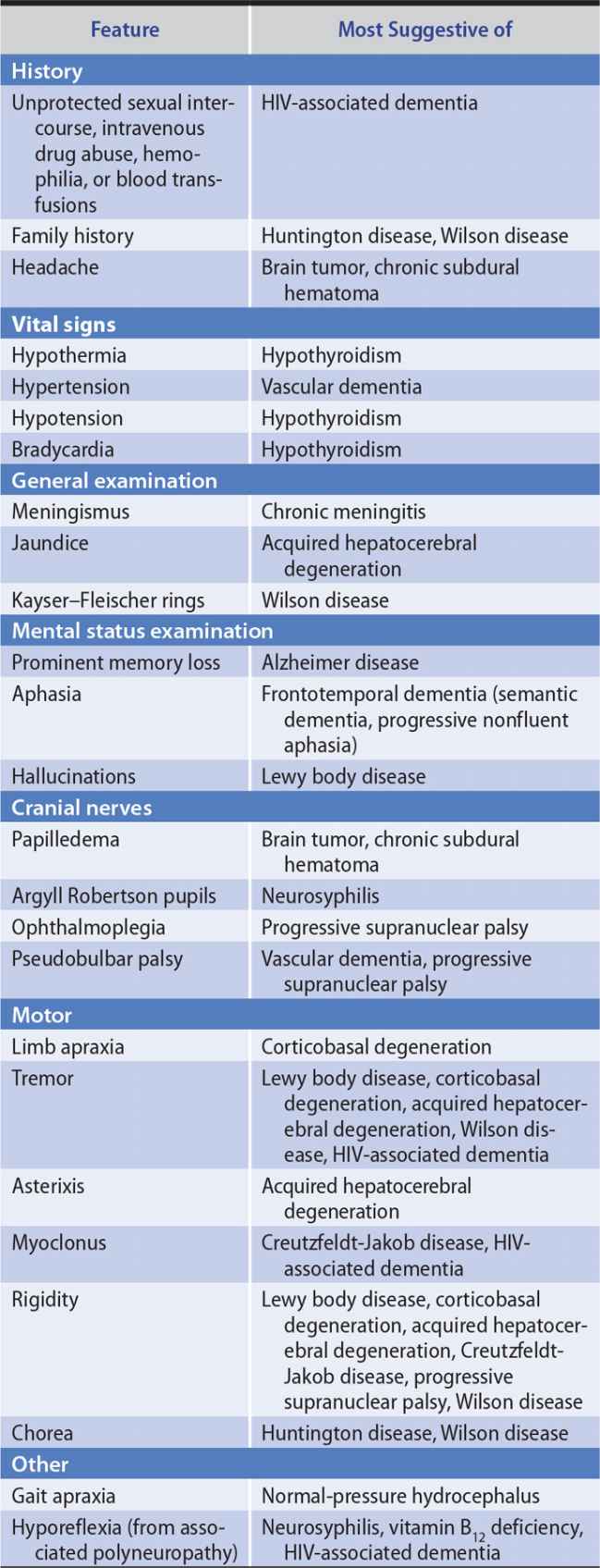
The general approach to obtaining a neurologic history is considered in Chapter 1, Neurologic History & Examination. Because dementia implies deterioration in cognitive ability, it is important to establish that the patient’s level of functioning has declined. Data that can help to establish the cause of dementia include the time course of deterioration; associated symptoms such as headache, gait disturbance, or incontinence; family history of a similar condition; concurrent medical illnesses; and the use of alcohol and therapeutic or recreational drugs (Table 5-3).
Table 5-3. Clinical Features Helpful in the Differential Diagnosis of Dementia.
GENERAL PHYSICAL EXAMINATION
The general physical examination can contribute to etiologic diagnosis when it reveals signs of a systemic disease responsible for dementia. Particularly helpful signs are listed in Table 5-3.
MENTAL STATUS EXAMINATION
The mental status examination (Table 5-4) helps to determine whether the level or the content of consciousness is impaired and whether cognitive dysfunction is global or circumscribed. A disorder of the level of consciousness is suggested by sleepiness, inattention, impairment of immediate recall, or disorientation regarding place or time. Abnormalities in these areas are unusual in dementia until the disorder is far advanced.
Table 5-4. Comprehensive Mental Status Examination.
Level of consciousness
Arousability
Orientation
Attention
Concentration
Language and speech
Comprehension
Repetition
Fluency
Naming
Reading
Writing
Calculation
Speech
Mood and behavior
Mood
Content of thought
Hallucinations
Delusions
Abstraction
Judgment
Memory
Immediate recall
Recent memory
Remote memory
Integrative sensory function
Astereognosis
Agraphesthesia
Two-point discrimination
Allesthesia
Extinction
Unilateral neglect and anosognosia
Disorders of spatial thought
Integrative motor function
Apraxia
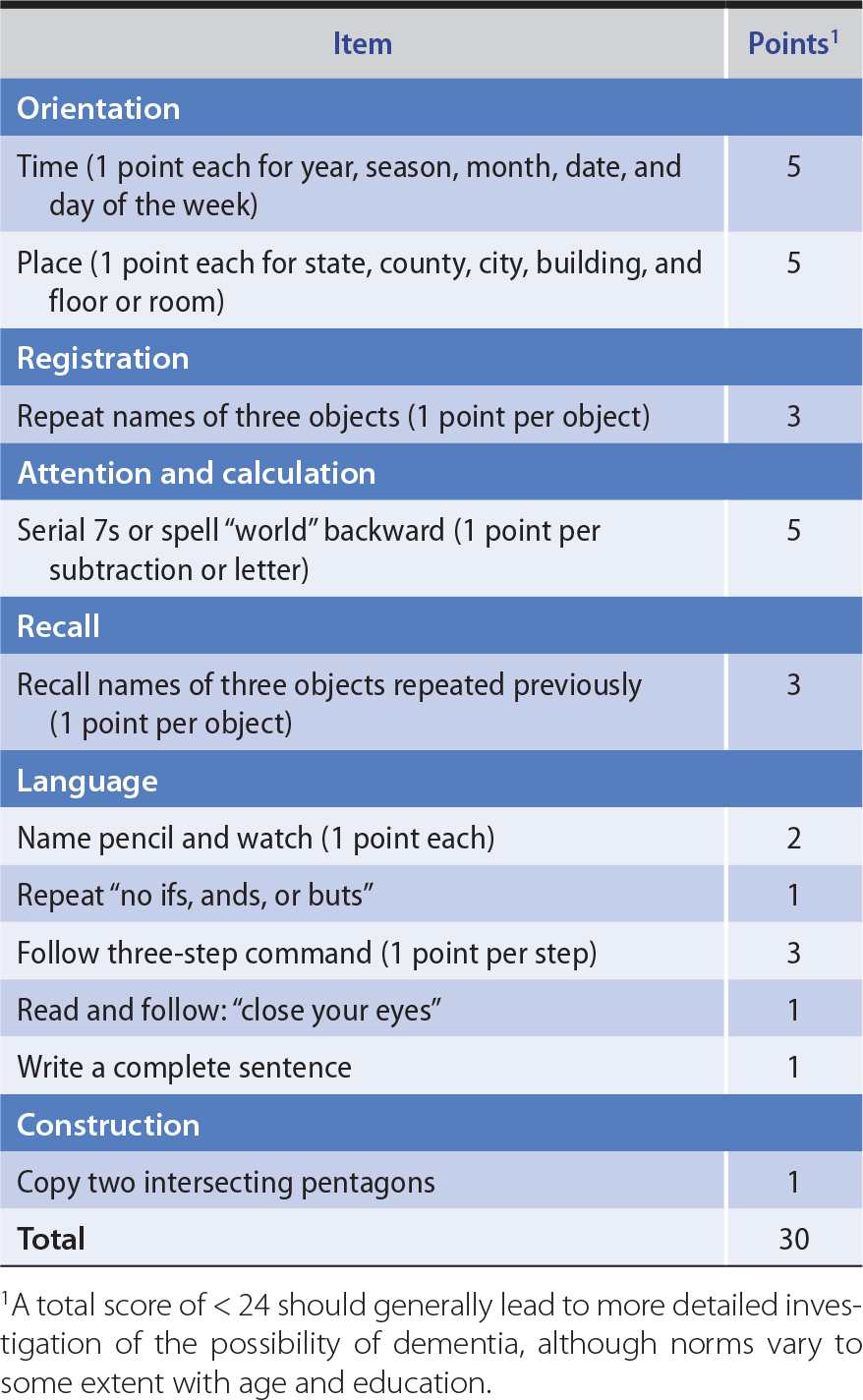
To determine the scope of the cognitive dysfunction (global or circumscribed), different spheres of cognition are tested in turn. These include memory, language, parietal lobe functions (pictorial construction, right–left discrimination, localization of objects in space), and frontal lobe or diffuse cerebral cortical functions (judgment, abstraction, thought content, the ability to perform previously learned acts). Multiple areas of cognitive function are impaired in dementia. The Montreal Cognitive Assessment (available at www.mocatest.org) and Minimental Status Examination (Table 5-5) provide useful bedside screening tests when dementia is suspected, but may not detect mild cognitive impairment.
Table 5-5. Minimental Status Examination.
Dementia from different causes may preferentially impair different spheres of cognition, and this can provide diagnostic clues. For example, Alzheimer disease affects memory disproportionately, whereas language function is often most impaired in frontotemporal dementia.
NEUROLOGIC EXAMINATION
Certain disorders that produce dementia also affect vision, coordination, or motor or sensory function. Detecting such associated neurologic abnormalities can help to establish an etiologic diagnosis. Neurologic signs suggesting causes of dementia are listed in Table 5-3.
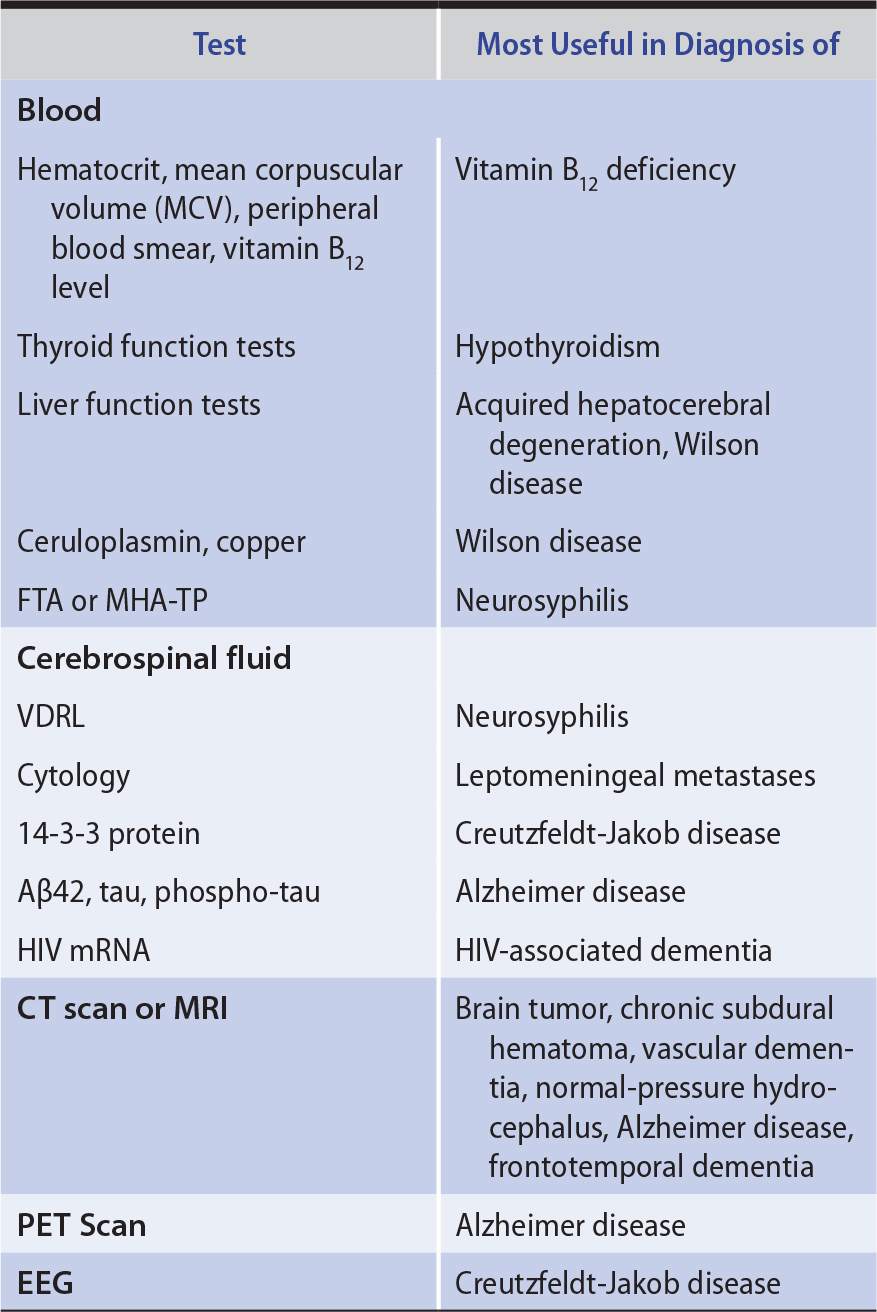
LABORATORY INVESTIGATIONS
Laboratory studies that can help to identify the cause of dementia are listed in Table 5-6.
Table 5-6. Laboratory Studies in Dementia.
DEMENTIA
DIFFERENTIAL DIAGNOSIS
 Common Causes of Dementia
Common Causes of Dementia
A wide variety of diseases can produce dementia, but only a few do so commonly. In its most typical presentation—with gradual cognitive decline in an elderly (≥65 years) patient—the most common causes of dementia are Alzheimer disease, vascular (formerly “multi-infarct”) dementia, frontotemporal dementia, Lewy body disease, and Parkinson disease (Figure 5-3). In many patients, neurodegeneration and vascular disease coexist as causes of dementia in the same patient. In addition, vascular factors may influence the risk or progression of neurodegenerative disease. Patients who present with dementia before age 45 years are much less likely to have Alzheimer disease or vascular dementia; in these patients, a wider range of neurodegenerative (Huntington disease, corticobasal degeneration), inflammatory (multiple sclerosis, systemic lupus erythematosus, vasculitis), and infective (prion disease) causes must be entertained. Dementia that progresses rapidly over weeks to months results most often from prion (Creutzfeldt–Jakob) disease.
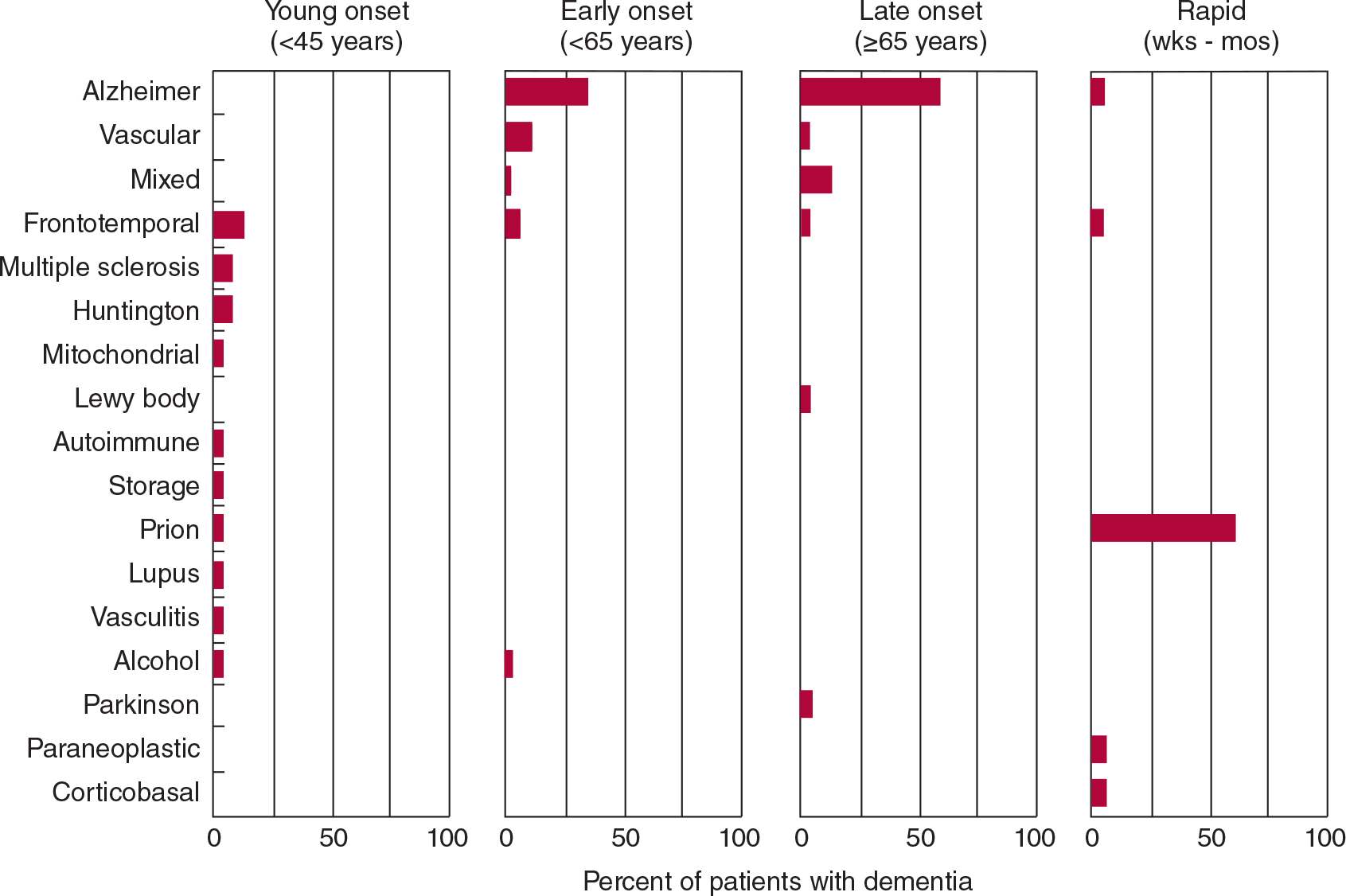
![]() Figure 5-3. Common causes of dementia based on age at presentation and rate of progression. “Mixed” refers to combined Alzheimer disease and vascular dementia. Totals of <100% reflect absence of an etiologic diagnosis in some cases. (Data from: Kelley BJ, Boeve BF, Josephs KA. Young-onset dementia: demographic and etiologic characteristics of 235 patients. Arch Neurol. 2008;65:1502-1508. Garre-Olmo J, Genís Batlle D, del Mar Fernández M, et al. Incidence and subtypes of early-onset dementia in a geographically defined general population. Neurology. 2010;75:1249-1255. Geschwind MD, Shu H, Haman A, Sejvar JJ, Miller BL. Rapidly progressive dementia. Ann Neurol. 2008;64:97-108.)
Figure 5-3. Common causes of dementia based on age at presentation and rate of progression. “Mixed” refers to combined Alzheimer disease and vascular dementia. Totals of <100% reflect absence of an etiologic diagnosis in some cases. (Data from: Kelley BJ, Boeve BF, Josephs KA. Young-onset dementia: demographic and etiologic characteristics of 235 patients. Arch Neurol. 2008;65:1502-1508. Garre-Olmo J, Genís Batlle D, del Mar Fernández M, et al. Incidence and subtypes of early-onset dementia in a geographically defined general population. Neurology. 2010;75:1249-1255. Geschwind MD, Shu H, Haman A, Sejvar JJ, Miller BL. Rapidly progressive dementia. Ann Neurol. 2008;64:97-108.)
 Other Causes of Dementia
Other Causes of Dementia
Reversible causes of dementia, such as normal-pressure hydrocephalus, intracranial mass lesions, vitamin B12 deficiency, hypothyroidism, and neurosyphilis, are rare. However, they are important to diagnose because treatment can arrest or reverse intellectual decline.
Diagnosing dementia caused by Huntington disease or other heritable disorders allows patients and their families to benefit from genetic counseling. If Creutzfeldt-Jakob disease or HIV-associated dementia is diagnosed, precautions can be instituted against transmission, and HIV disease can be treated with antiretroviral drugs. Progressive multifocal leukoencephalopathy may indicate underlying immunosuppression from HIV infection, lymphoma, or leukemia and may thereby bring these disorders to attention.
Approximately 15% of patients referred for evaluation of possible dementia instead have other disorders (pseudodementias), such as depression. Depression in this setting is important to identify because it is readily treatable. Drug intoxication, often cited as a cause of dementia in the elderly, actually produces an acute confusional state, rather than dementia.
NEURODEGENERATIVE PROTEINOPATHIES
In several neurodegenerative diseases, the production of misfolded proteins and their association to form insoluble aggregates appears to play an important role in pathogenesis (Table 5-7). These abnormal proteins can arise from either genetic or acquired modifications, and their pathologic effects may result from loss of normal protein function, gain of a toxic function, or a combination of these factors. Protein aggregation may be a mechanism for sequestering proteins that the cell’s proteolytic machinery cannot process, but protein aggregates may also exert adverse effects on the cell, such as by interfering with axonal transport.
Table 5-7. Neurodegenerative Proteinopathies.
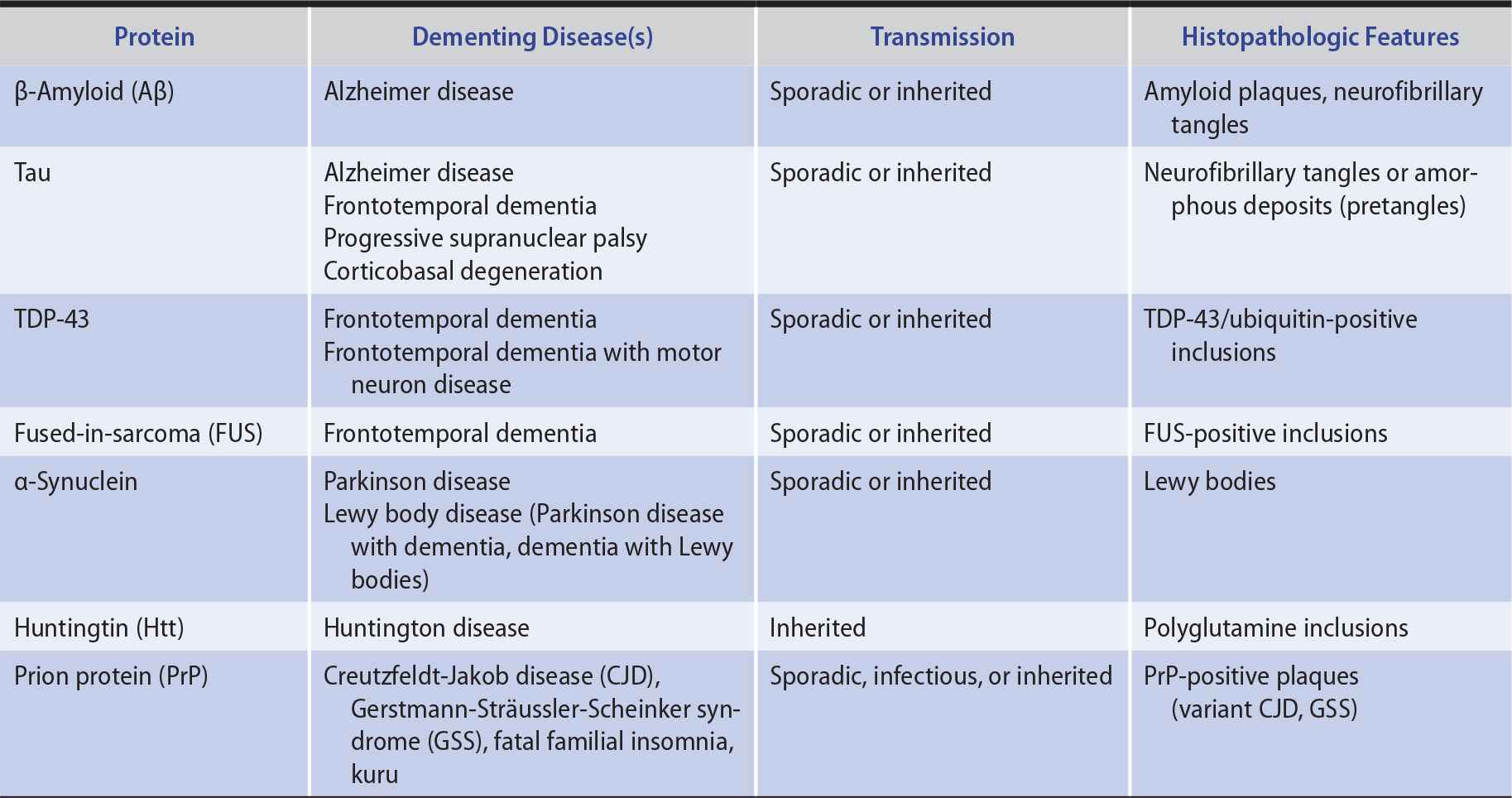
Except in rare inherited or infectious cases, the underlying cause of neurodegenerative proteinopathies is unknown. However, these diseases share several features. In addition to protein misfolding and aggregation, which sometimes produces characteristic histopathologic findings (see Table 5-7), these diseases may be associated with cell-to-cell prionic transmission (Figure 5-4), which allows them to spread through the nervous system to produce characteristic anatomic patterns of involvement. Evidence for this mechanism includes the finding that fetal tissue grafted into the brains of patients with Parkinson disease develops the same abnormal protein aggregates found in the recipient’s brain.
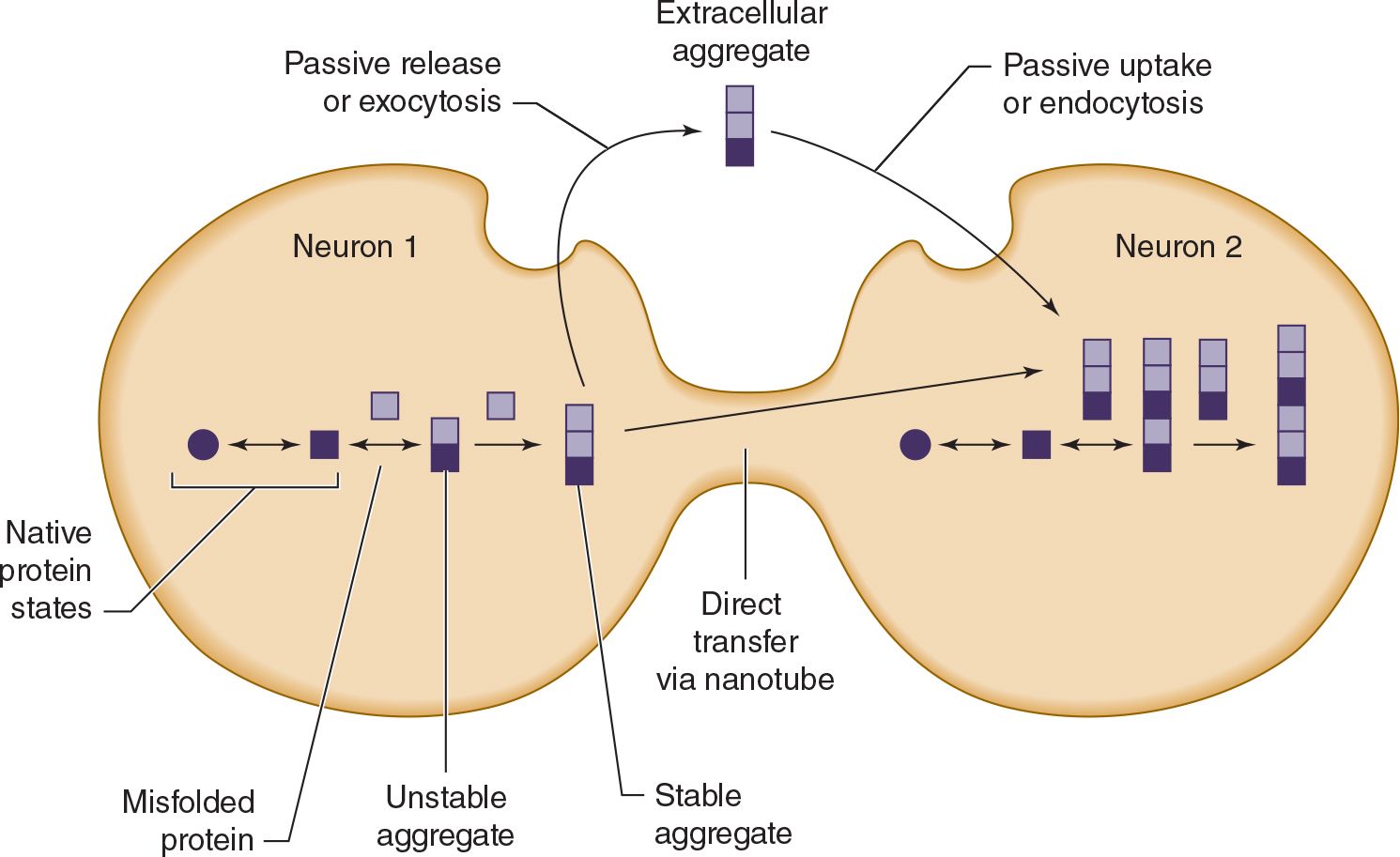
![]() Figure 5-4. Cell-to-cell (prionic) transmission of neurodegenerative proteinopathies. Abnormal proteins associated with neurodegenerative disease may misfold, leading to the formation of protein aggregates; misfolded proteins, protein aggregates, or both may be toxic and contribute to neuronal dysfunction. In addition, toxic protein aggregates may be transferred between cells to propagate the disease.
Figure 5-4. Cell-to-cell (prionic) transmission of neurodegenerative proteinopathies. Abnormal proteins associated with neurodegenerative disease may misfold, leading to the formation of protein aggregates; misfolded proteins, protein aggregates, or both may be toxic and contribute to neuronal dysfunction. In addition, toxic protein aggregates may be transferred between cells to propagate the disease.
ALZHEIMER DISEASE
 Epidemiology
Epidemiology
Alzheimer disease is the most common cause of dementia, accounting in whole or part for an estimated 60% to 70% of cases. Alzheimer disease affects approximately 15% of individuals of age 65 years or older and approximately 45% of those age 85 years or over. Its prevalence is >5 million cases in the United States and approximately 30 million cases worldwide. Men and women are affected with equal frequency, when adjusted for age. However, because women live longer, they account for approximately two-thirds of Alzheimer patients.
 Pathology
Pathology
Alzheimer disease is defined by characteristic histopathologic features, especially neuritic (senile) plaques and neurofibrillary tangles. Neuritic plaques are extracellular deposits that contain β-amyloid (Aβ) and other proteins, including presenilin 1, presenilin 2, α1-antichymotrypsin, apolipoprotein E, α2-macroglobulin, and ubiquitin. Plaques may also be found in cerebral and meningeal blood-vessel walls, producing cerebral amyloid angiopathy. Neurofibrillary tangles are intracellular deposits containing hyperphosphorylated tau (a microtubule-associated protein) and ubiquitin.
 Etiology
Etiology
Alzheimer disease is a progressive, degenerative disorder that is caused by a genetic defect in rare cases (see later), but is usually sporadic and of unknown cause. Abnormal metabolism, deposition, or clearance of two proteins—Aβ and tau—appears to be closely linked to pathogenesis.
 Pathogenesis
Pathogenesis
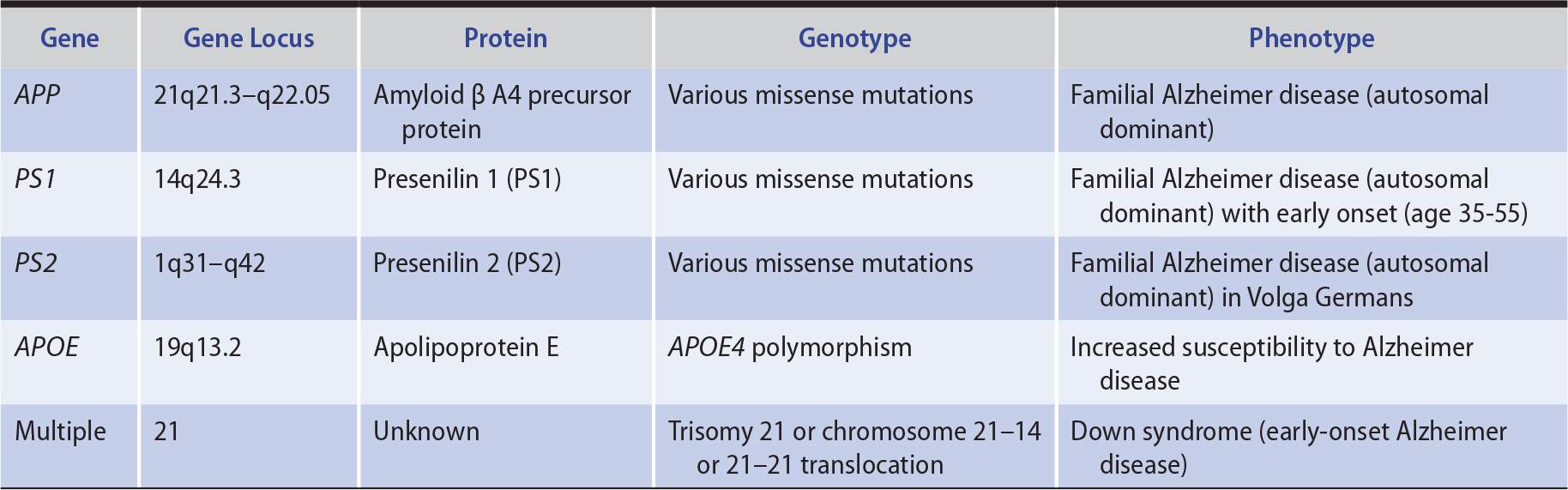
1. Genetics—In approximately 1% of patients, Alzheimer disease is a familial disorder that results from a mutation in one of three functionally related membrane proteins (Table 5-8): β-amyloid precursor protein (APP), presenilin 1 (PS1), or presenilin 2 (PS2). Onset of the disease in these patients is typically between the ages of 30 and 60 years. Patients with Down syndrome (trisomy 21) also develop early Alzheimer disease (mean onset at age 50 years), which is thought to be related to an extra copy of the APP gene, located on chromosome 21. Although the cause of sporadic Alzheimer disease is unknown, the gene defects in familial Alzheimer disease provide clues, implicating both APP, a protein with neurotrophic properties, and the presenilins, which are involved in APP metabolism.
The risk of Alzheimer disease is also influenced by the inheritance pattern of apolipoprotein E (APOE) gene isoforms ε2, ε3, and ε4. Risk increases about three-fold with a single apolipoprotein Eε4 (APOE4) allele and about 12-fold with two copies of APOE4; each copy of APOE4 also lowers the age at onset by about 5 years. In contrast to APOE4, APOE2 appears to confer relative protection from Alzheimer disease. The mechanism through which APOE genotype modifies susceptibility to Alzheimer disease is unknown, but may involve binding of the APOE protein to Aβ.
2. Aβ and neuritic plaques—Aβ is the principal constituent of neuritic plaques and is also deposited in cerebral and meningeal blood vessels in Alzheimer disease. Aβ is a 38- to 43-amino acid peptide produced by proteolytic cleavage of the transmembrane protein, APP (Figure 5-5). Normal processing of APP involves its cleavage by the enzyme α-secretase, which does not produce Aβ, and by β-secretase (BACE; β-site APP cleaving enzyme) and γ-secretase, yielding primarily a 40-amino acid fragment (Aβ40), which is secreted and cleared from the brain. In Alzheimer disease, a disproportionate amount of Aβ42, a longer form of the molecule with an increased tendency to aggregate, is produced. Presenilins 1 and 2 contribute to γ-secretase activity.
Evidence for a causal role of Aβ in Alzheimer disease includes the involvement of APP mutations in some familial cases and the neurotoxicity of Aβ under some circumstances. However, there is a poor correlation between the extent of amyloid plaque deposition in the brain and the severity of dementia in Alzheimer disease. One explanation for this disparity is that soluble Aβ oligomers, rather than insoluble plaques, may be the toxic agent. Another possibility is that Aβ aggregation produces Alzheimer disease indirectly, such as by promoting the formation of tau-containing neurofibrillary tangles.
3. Tau and neurofibrillary tangles—Tau is a cytoplasmic protein that binds to tubulin and stabilizes microtubules, cytoskeletal structures that help maintain cell structure and facilitate intracellular transport. In Alzheimer disease and other tauopathies, tau becomes hyperphosphorylated and dissociates from microtubules; the microtubules disassemble and hyperphosphorylated tau aggregates to form neurofibrillary tangles (Figure 5-6). How this leads to impaired neuronal function is unknown, but may involve a defect in axonal transport. A causal role for tau pathology in Alzheimer disease is supported by the observations that the abundance of neurofibrillary tangles correlates well with disease severity and that other tauopathies (eg, frontotemporal dementia), in which Aβ processing is normal, can also produce dementia.
4. Synaptic dysfunction—Alzheimer disease is accompanied by early dysfunction and later loss of synapses, prominently affecting excitatory transmission in hippocampus and cerebral cortex. These changes may contribute to memory loss.
5. Neuronal loss and brain atrophy—Certain neuronal populations are preferentially lost in Alzheimer disease, including glutamatergic neurons in the entorhinal cortex and the CA1 sector of hippocampus, as well as cholinergic neurons in the basal forebrain. Focal brain atrophy is seen in the affected areas.
6. Vascular involvement—The extent to which vascular pathology may contribute to Alzheimer disease is controversial. Evidence for such a connection includes the overlap between risk factors for vascular disease and Alzheimer disease (including APOE genotype) and the involvement of blood vessels in amyloid pathology.
Table 5-8. Principal Genes Implicated in Alzheimer Disease.
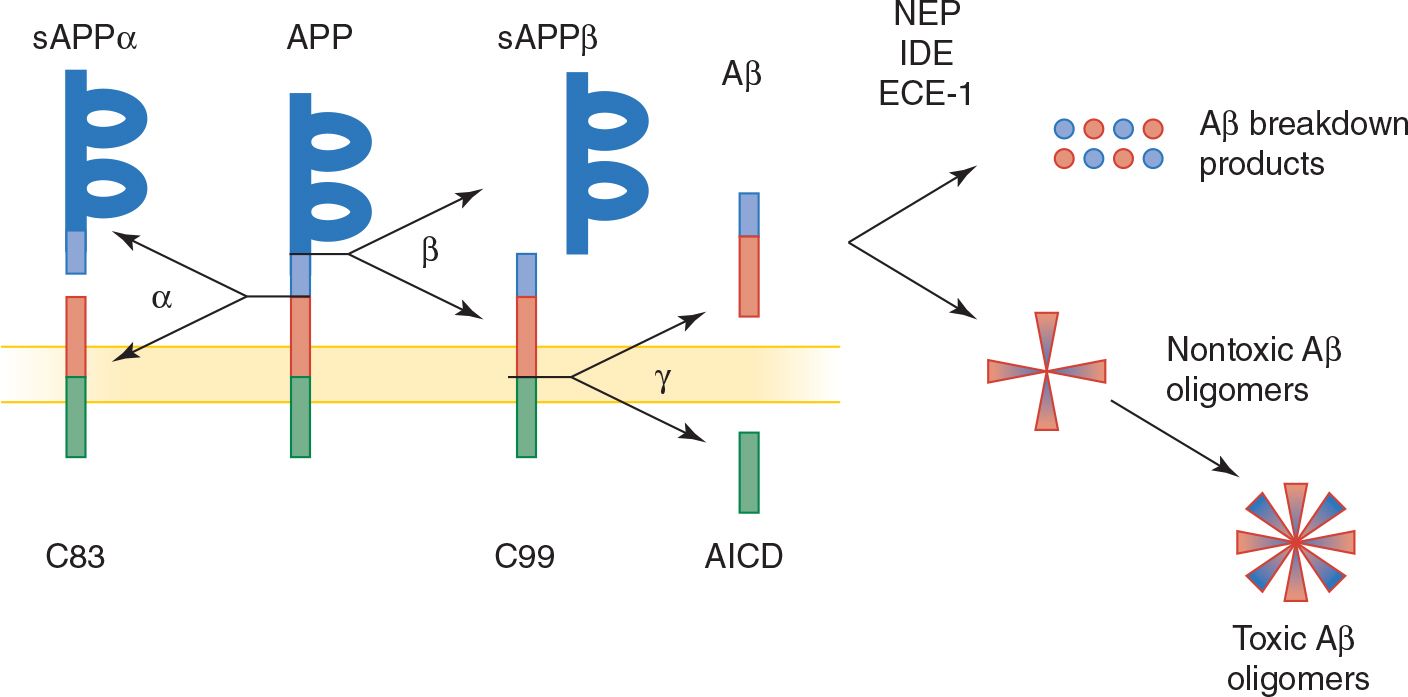
![]() Figure 5-5. Normal and pathologic (amyloidogenic) processing of APP and Aβ. APP, a membrane-spanning protein, is normally cleaved by α-secretase (α), or by β-secretase (β) and then γ-secretase (γ, a protein complex that includes presenilin 1 or 2, nicastrin, anterior pharynx defective 1 homolog [APH1], and presenilin enhancer 2 [PEN2]) to generate β-amyloid (Aβ), a secreted protein. APP mutations associated with familial Alzheimer disease shift Aβ production from a nontoxic 40-amino acid to a toxic (amyloidogenic) 42-amino acid form, which has a greater tendency to form amyloid deposits. Aβ normally undergoes enzymatic breakdown (by neprilysin [NEP], insulin-degrading enzyme [IDE], or endothelin-converging enzyme [ECE-1]) and clearance from the brain. However, it can also aggregate to form oligomers of increasing size, which are thought to be neurotoxic. sAPP, soluble APP; C83 and C99, C-terminal fragments of APP; AICD, APP intracellular domain.
Figure 5-5. Normal and pathologic (amyloidogenic) processing of APP and Aβ. APP, a membrane-spanning protein, is normally cleaved by α-secretase (α), or by β-secretase (β) and then γ-secretase (γ, a protein complex that includes presenilin 1 or 2, nicastrin, anterior pharynx defective 1 homolog [APH1], and presenilin enhancer 2 [PEN2]) to generate β-amyloid (Aβ), a secreted protein. APP mutations associated with familial Alzheimer disease shift Aβ production from a nontoxic 40-amino acid to a toxic (amyloidogenic) 42-amino acid form, which has a greater tendency to form amyloid deposits. Aβ normally undergoes enzymatic breakdown (by neprilysin [NEP], insulin-degrading enzyme [IDE], or endothelin-converging enzyme [ECE-1]) and clearance from the brain. However, it can also aggregate to form oligomers of increasing size, which are thought to be neurotoxic. sAPP, soluble APP; C83 and C99, C-terminal fragments of APP; AICD, APP intracellular domain.
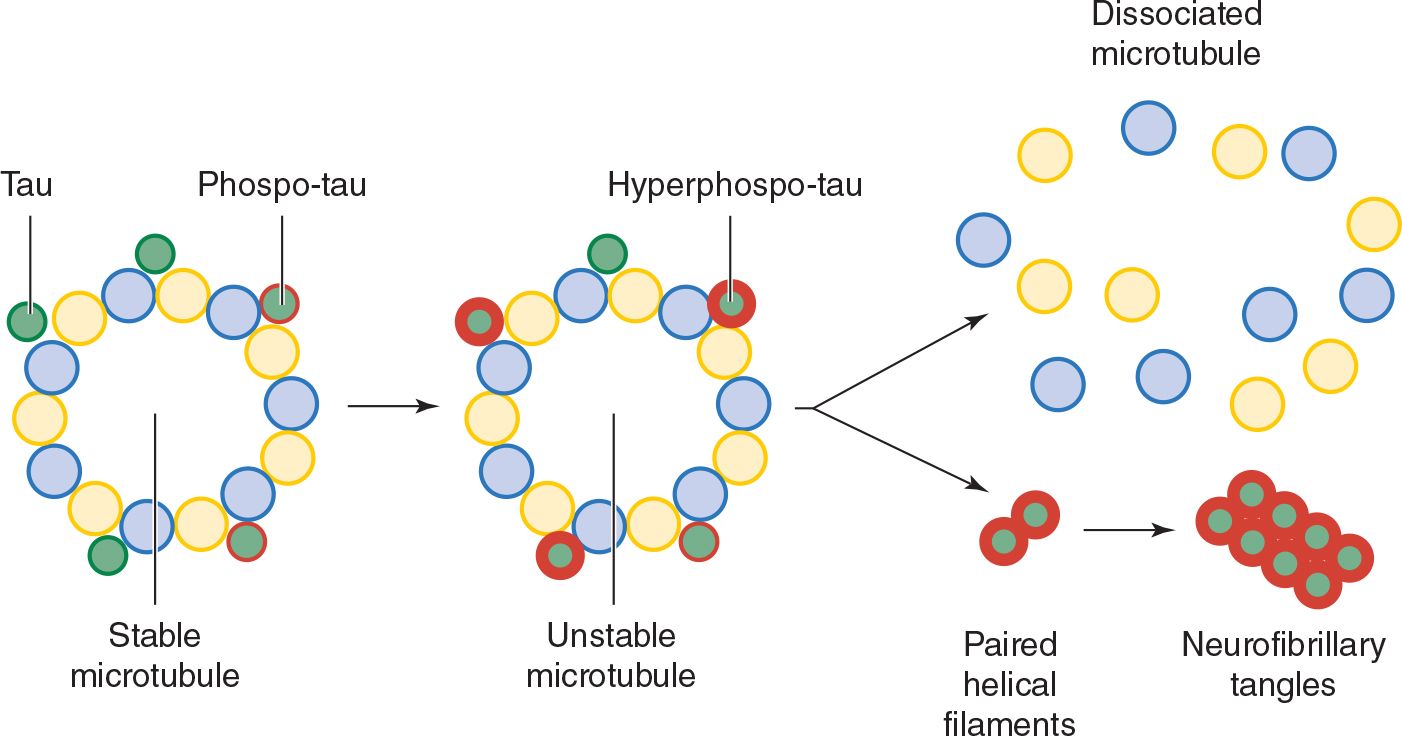
![]() Figure 5-6. Tau hyperphosphorylation and neurofibrillary tangle formation.
Figure 5-6. Tau hyperphosphorylation and neurofibrillary tangle formation.
 Risk Factors
Risk Factors
The factors most conclusively associated with increased risk for Alzheimer disease are increasing age, female sex, and APOE4 genotype. Other factors that have been implicated in some studies include family history of Alzheimer disease, depression, low educational level, smoking, diabetes, hypertension, and fatty diet. Besides APOE4, several other genes that modify risk have been identified, but the magnitude of their effects is small.
Definitive evidence that the risk of Alzheimer disease can be reduced by diet, drugs, or lifestyle changes is lacking. However, cognitive engagement, physical activity, a low-fat and vegetable-rich diet, and light to moderate alcohol intake are all supported by some data.
 Clinical Findings
Clinical Findings
The clinical progression of Alzheimer disease is thought to comprise a presymptomatic phase of up to about 10 years characterized by the deposition of amyloid plaques, followed by a symptomatic phase of up to about 10 years, during which tangle formation occurs (Figure 5-7).
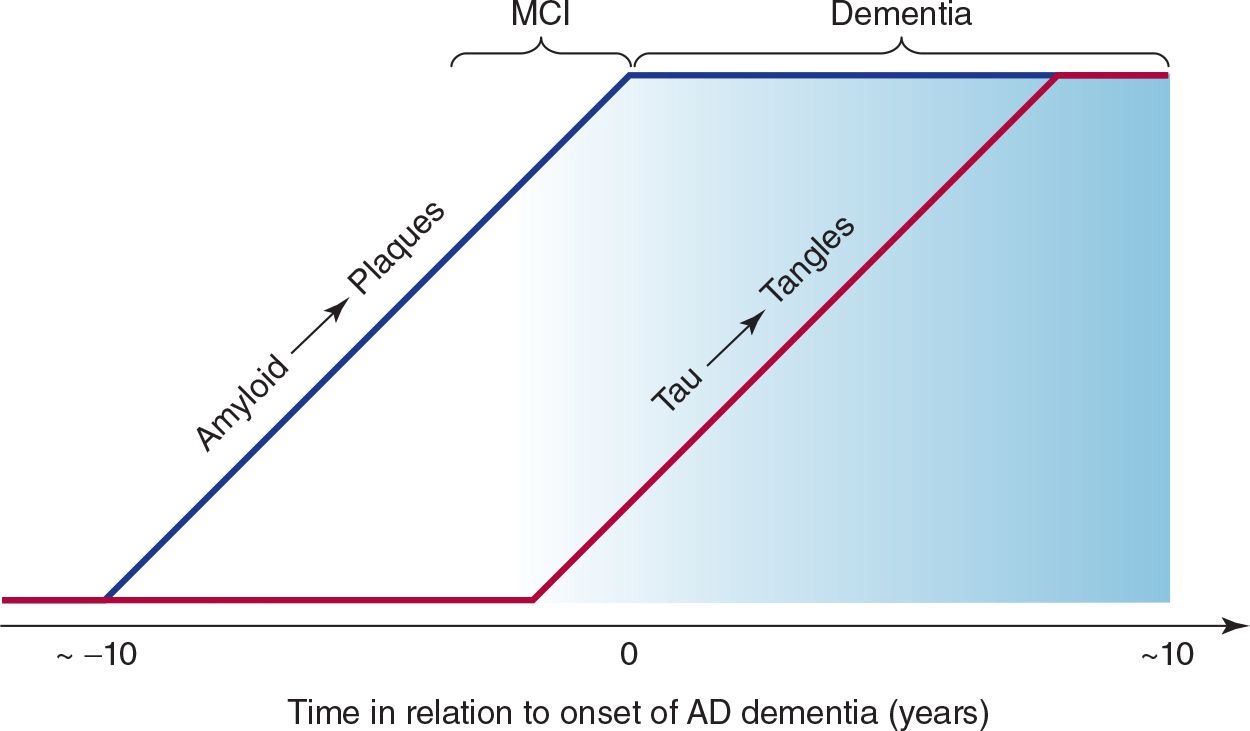
![]() Figure 5-7. Relationship between plaques, tangles, and clinical progression of Alzheimer disease (AD). MCI, mild cognitive impairment.
Figure 5-7. Relationship between plaques, tangles, and clinical progression of Alzheimer disease (AD). MCI, mild cognitive impairment.
1. Early manifestations—The term mild cognitive impairment (MCI) is sometimes used to describe the early phase of cognitive decline observed in patients who later receive a diagnosis of Alzheimer disease. Impairment of recent memory is typically the first sign of Alzheimer disease and may be noticed only by family members. As the memory disorder progresses over months to several years, the patient becomes disoriented to time and then to place. Aphasia, anomia, and acalculia may develop, forcing the patient to leave work or give up the management of family finances. The depression apparent in the earlier stages of the disorder may give way to an agitated, restless state. Apraxias and visuospatial disorientation ensue, causing the patient to become lost easily. Primitive reflexes are commonly found. A frontal lobe gait disorder may become apparent, with short, slow, shuffling steps, flexed posture, wide base, and difficulty in initiating walking.
2. Late manifestations—In the late stages, previously preserved social graces are lost, and psychiatric symptoms, including psychosis with paranoia, hallucinations, or delusions, may be prominent. Seizures occur in some cases. Examination at this stage may show rigidity and bradykinesia. Rare and usually late features of the disease include myoclonus, incontinence, spasticity, extensor plantar responses, and hemiparesis. Mutism, incontinence, and a bedridden state are terminal manifestations. Eating problems, febrile episodes, dyspnea, pneumonia, and pain are frequent complications in the final months of life, and death typically occurs from 5 to 10 years after the onset of symptoms.
 Investigative Studies
Investigative Studies
Laboratory investigations can exclude other disorders, including reversible or otherwise treatable conditions. Cognitive testing may also be useful in helping to distinguish between Alzheimer disease and other causes of dementia. In patients with Alzheimer disease, the CT scan or MRI often shows cortical (especially medial temporal lobe) atrophy and enlarged ventricles, but such changes are nonspecific. Positron emission tomography (PET) scanning may reveal hypometabolism and hypoperfusion in the temporal and parietal lobes. More specific PET tracers include [18F]FDDNP, which labels amyloid plaques and neurofibrillary tangles, and [11C] Pittsburgh compound B (PIB), which binds to amyloid plaques. Cerebrospinal fluid (CSF) levels of Aβ42, tau, and phospho-tau have also been found useful as biomarkers of Alzheimer disease.
 Differential Diagnosis
Differential Diagnosis
Early Alzheimer disease may resemble depression or pure memory disorders such as the Korsakoff amnestic syndrome (see later discussion). More advanced Alzheimer disease must be distinguished from Lewy body dementia, vascular dementia, Creutzfeldt-Jakob disease, and other dementing disorders (see later discussion).
 Treatment
Treatment
No currently available treatment has been shown to reverse existing deficits or to arrest disease progression. However, memantine (Table 5-9), an NMDA-type glutamate receptor antagonist drug, may produce modest improvement in patients with moderate or severe Alzheimer disease.
Table 5-9. Drugs Used in the Treatment of Alzheimer Disease.
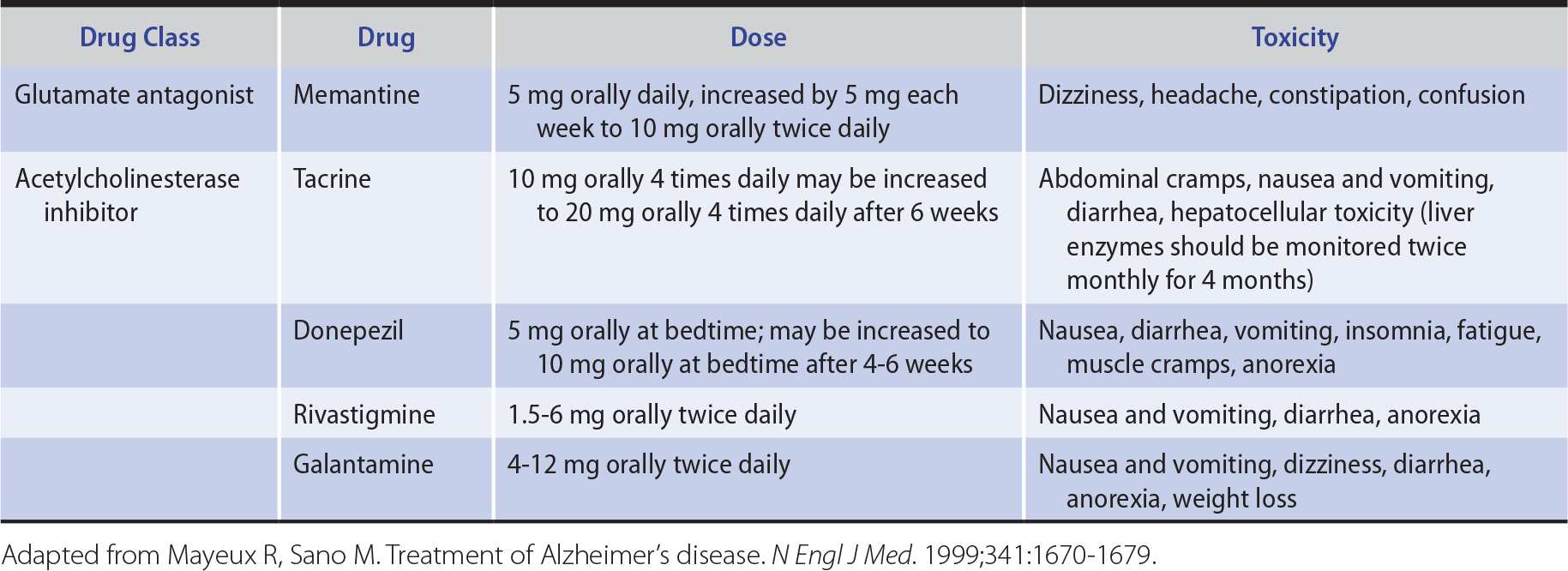
Because cholinergic neuronal pathways degenerate and choline acetyltransferase is depleted in the brains of patients with Alzheimer disease, cholinergic replacement therapy has also been used for symptomatic treatment of cognitive dysfunction (see Table 5-9). Acetylcholinesterase inhibitors, including tacrine, donepezil, rivastigmine, and galantamine, have all been shown to produce small improvements in tests of cognitive function. Side effects include nausea and vomiting, diarrhea, and dizziness; tacrine also elevates serum transaminase levels. The better side-effect profile of donepezil and its once-daily dosage schedule are advantageous.
Experimental treatments under investigation include vaccines directed against β-amyloid, secretase inhibitors, and metal chelators. Antipsychotic drugs, antidepressants, and anxiolytics may be useful in controlling behavioral disturbances associated with Alzheimer disease. However, evidence for their effectiveness is sparse, and in some cases (risperidone, olanzapine) their use is associated with an increased incidence of stroke in elderly patients.
 Prognosis
Prognosis
Early in the course of the disease, patients can usually remain at home and continue social, recreational, and limited professional activities. Early diagnosis can allow patients time to plan orderly retirement from work, to arrange for management of their finances, and to discuss with physicians and family members the management of future medical problems. Patients in advanced stages of the disease may require care in a nursing facility and the use of psychoactive medications. These patients must be protected and prevented from injuring themselves and their families by injudicious actions or decisions. Death from inanition or infection generally occurs 5 to 10 years after the first symptoms.



