Dementia, delirium, and other neuropsychiatric disorders
Symptoms associated with regional brain pathology
Assessment of the ‘neuropsychiatric patient’
Amnesia and amnestic syndromes
Other neuropsychiatric syndromes
Secondary or symptomatic neuropsychiatric syndromes
Introduction
Neuropsychiatry comprises psychiatric disorders that arise from demonstrable abnormalities of brain structure and function. Cognitive impairments are the most prominent feature, especially in dementia and delirium, but behavioural and emotional disturbances are also common, and may be the sole manifestations.
The term neuropsychiatry is sometimes used interchangeably with organic psychiatry (see David, 2009a). However, the latter category is broader, including psychiatric disorders that arise from general medical disorders with their basis outside the brain (e.g. endocrine and metabolic disorders). These disorders are covered in Chapter 15. Moreover, the term organic has the fundamental problem that it wrongly implies that other psychiatric disorders do not have any such basis (see Chapter 2, p. 26); neuroscience increasingly shows the falsity of this dichotomy. Finally, both terminologies run the risk that psychological and social factors will be neglected because the disorder is considered to be ‘physical.’
In this chapter we cover the range of disorders conventionally considered under the heading of neuropsychiatry, which include:
• delirium—acute, generalized cognitive impairment in the setting of altered consciousness
• dementia—chronic, generalized cognitive impairment in clear consciousness. As with delirium, the syndrome of dementia can be caused by many separate disease processes (this chapter covers the clinical features and aetiology of dementia; its treatment and management are covered in Chapter 18)
• amnestic (or amnesic) syndromes—circumscribed deficits in memory
• epilepsy
• head injury
• other neuropsychiatric disorders, including focal cerebral syndromes, infections, tumours, and multiple sclerosis
• secondary or symptomatic neuropsychiatric disorders—disorders such as depression and anxiety which in particular cases can be attributed directly to a neuropsychiatric cause (e.g. psychosis due to cerebral vasculitis). (Psychiatric disorders secondary to diseases elsewhere in the body are covered in Chapter 15.)
Classification
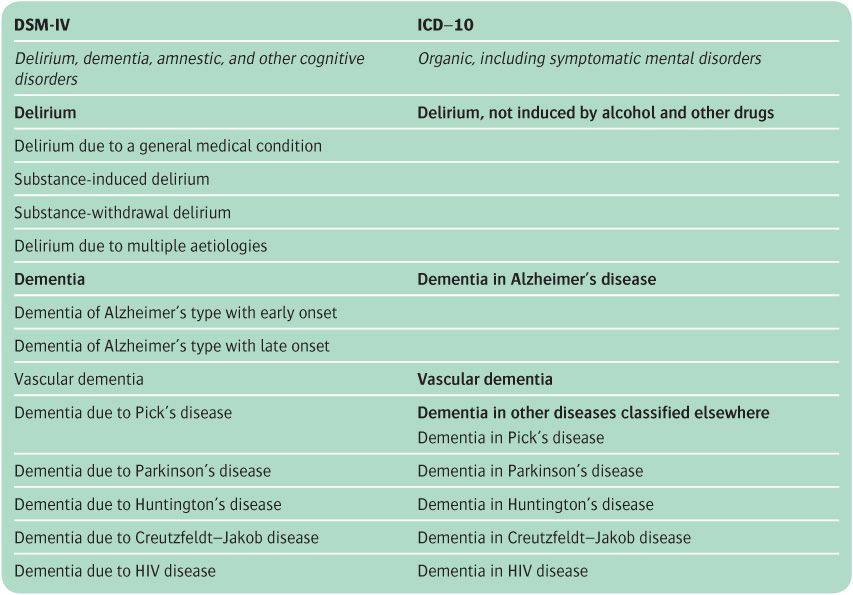
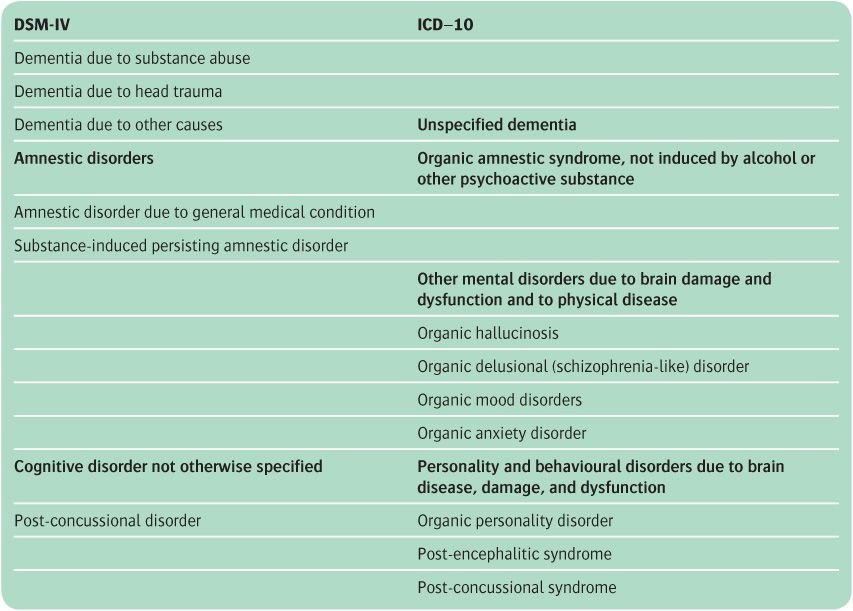
In ICD-10, organic psychiatric disorders are termed organic, including symptomatic, mental disorders, and in DSM-IV they are categorized as delirium, dementia, amnestic and other cognitive disorders. The two classifications are compared in Table 13.1. The main differences can be summarized as follows.
• The omission of ‘organic’ from the section title in DSM-IV led to rearrangement of the classification of some conditions formerly grouped under the heading ‘organic.’ Thus major depression with organic aetiology is classified under mood disorders either as secondary to a general medical condition, or as substance induced. As a result of these changes, DSM-IV avoided the problems within ICD-10 of the definition of the terms organic, symptomatic, and secondary (Spitzer et al., 1992).
• In both classifications the specific medical conditions causing cognitive disorder can be coded in addition to the latter disorder. In DSM-IV, this additional code is recorded on Axis III.
• In ICD-10, the section on organic disorder includes subcategories for mental disorders due to brain damage and dysfunction and to physical disease, and for personality and behavioural factors due to brain disease, damage, and dysfunction (e.g. ‘organic anxiety disorder, thyrotoxicosis’). In DSM-IV these conditions are classified under the relevant psychiatric disorder, with the addition of a code to indicate that the disorder is secondary to a medical condition.
• DSM-IV includes categories of substance-induced delirium, dementia, and amnestic disorders. In ICD-10 these conditions are recorded in the section on mental and behavioural disorders due to psychoactive substance abuse. Thus, for example, amnestic syndrome is coded in ICD-10 in this section, but amnestic syndrome due to alcohol (Korsakov’s syndrome) is classified as a psychoactive substance abuse disorder.
Table 13.1 Classification of organic mental disorders
Symptoms associated with regional brain pathology
Before considering the various syndromes, it is helpful to consider the characteristic features associated with lesions in different regions of the brain, and the neuro-anatomical basis of memory. Knowledge of the regional affiliation of neurological and psychopathological findings is relevant when attempting to localize neuropsychiatric conditions. However, the clinical features are not diagnostically specific, and the clinico-pathological correlations are often modest rather than strong. For a review of this subject, see David (2009a).
Frontal lobe
The frontal lobes, together with their reciprocal connections to other cortical and subcortical regions, have a crucial role in personality and judgement. Patients with a frontal lobe syndrome may present with a variety of clinical syndromes.
• They may be disinhibited, overfamiliar, tactless, and garrulous, make fatuous jokes and puns (Witzelsucht), commit errors of judgement and sexual indiscretions, and disregard the feelings of others.
• They may appear inert (abulic) and apathetic, with a paucity of spontaneous speech, movement, and emotional expressions.
• They may engage in obsessive, ritualistic behaviours, with perseveration of thought and gesture.
Measures of formal intelligence are generally unimpaired in frontal lobe disease. However, there may be difficulties in abstract reasoning (e.g. ‘How are glass and ice different?’) and cognitive estimates are typically inaccurate but precise (e.g. ‘364 miles from London to New York’). Concentration and attention are reduced, and insight is often markedly impaired. Verbal fluency, assessed using word generation by letter (e.g. number of words beginning with ‘s’ in one minute) and category (e.g. number of animals), is reduced, and unusual (low-frequency) examples may be volunteered. The patient has difficulty switching between tasks (perseveration), carrying out sequenced movements, and understanding rules. Utilization behaviour (e.g. donning several pairs of spectacles) may be evident.
Posterior extension of a dominant frontal lobe lesion may involve Broca’s area and produce an expressive (non-fluent) dysphasia. Encroachment on the motor cortex or deep projections may result in a contralateral hemiparesis. Other signs may include ipsilateral optic atrophy or anosmia, a grasp or other primitive reflexes and, if the process is bilateral or in the midline, incontinence of urine.
Parietal lobe
Lesions of the parietal lobe may cause various neuropsychological disturbances which are easily mistaken for conversion disorder (see p. 394). Involvement of the non-dominant parietal lobe characteristically gives rise to visuospatial difficulties, with neglect of contralateral space, and constructional and dressing apraxias. Lesions of the dominant lobe may be associated with receptive dysphasia, limb apraxia, body image disorders, right–left disorientation, dyscalculia, finger agnosia, and agraphia. Other signs may include contralateral sensory loss, astereognosis and agraphaesthesia, and (with more extensive lesions) a contralateral hemiparesis or homonymous inferior quadrantanopia.
Persistent unawareness of neurological deficit (anosognosia) is not uncommon, especially with non-dominant parietal lesions. In extreme cases, the patient may deny that a paretic limb belongs to him. This should be distinguished from denial due to a psychological unwillingness to recognize disability and its consequences.
Temporal lobe
The temporolimbic syndromes are characterized by complex and wide-ranging neuropsychiatric clinical pictures. There may be personality change resembling that of frontal lobe lesions, but more often accompanied by specific cognitive deficits and neurological signs. The relatively florid behavioural disturbances that characterize the frontotemporal dementias reflect the combined temporal and frontal involvement, and their interconnections.
Unilateral medial temporal lobe lesions, especially those involving the hippocampus, produce lateralizing memory deficits—left hippocampal damage impairs verbal memory (and semantic impairment and fluent dysphasia), whereas right hippocampal damage affects non-verbal (spatial) aspects of memory. Some evidence also suggests that left medial temporal lobe lesions are more likely to produce psychotic symptoms, and right-sided lesions produce affective ones.
Occipital lobe
Occipital lobe lesions rarely present to psychiatrists, but they may cause disturbances of visual processing which are easily misinterpreted as being of psychological origin. Such phenomena occasionally accompany migraine or occipital lobe seizures. Complex visual hallucinations may occur with lesions involving visual association areas, sometimes referred to a hemianopic field. These include multiple visual images (polyopia), persistent aftertraces of the features of an image (visual perseveration or palinopsia), and distortions of the visual scene (metamorphopsia). Lesions that impinge anteriorly on the parietal or temporal lobes may produce visual disorientation (inability to localize objects in space under visual guidance) with asimultagnosia (difficulty in perceiving the visual scene as a unity), or prosopagnosia (inability to recognize familiar faces). In patients with suspected occipital lobe pathology, the visual fields should be mapped using perimetry, and neuropsychological tests performed to delineate visual agnosias and other higher-order derangements of visual processing. Some patients who are blind due to occipital lobe damage deny that they are blind (Anton’s syndrome).
Corpus callosum
Corpus callosum lesions (classically, the ‘butterfly glioma’) typically extend laterally into both hemispheres. They then produce a picture of severe and rapid intellectual deterioration, with localized neurological signs varying with the degree and direction of extension into adjacent structures. Pure callosal lesions (usually iatrogenic, following surgery for intractable epilepsy) can be surprisingly difficult to identify, and require specialized neuropsychological testing to expose a ‘disconnection syndrome’, reflecting disruption of interhemispheric communication. These unique ‘split-brain’ patients raise intriguing questions concerning the mechanisms that normally bind the two hemispheres together to generate a consistent, unitary sense of the self (Gazzaniga, 2000). Callosal degeneration is a hallmark of the rare Marchiafava–Bignami syndrome, which is seen in severe alcohol dependence.
Subcortical structures and cortico-subcortical circuits
The regional cortical associations with cognitive, affective, and behavioural features reflect the classic ‘locationist’ approach to neurology and neuropsychiatry. Increasingly, this is being complemented, if not replaced, by a ‘connectionist’ approach, in which emphasis is placed on distributed neural systems in which cortical regions are linked with subcortical structures, and with the white matter pathways that connect them.
Mesulam (1998) described five networks:
• a right-hemisphere spatial awareness network including the posterior parietal cortex and frontal eye fields
• a left-hemisphere language network including Broca’s and Wernicke’s areas
• a memory–emotion network including the hippocampus, amygdala and cingulate cortex
• a working memory–executive function network including the prefrontal cortex and posterior parietal cortex
• a face and object recognition network in the temporoparietal and temporo-occipital cortex.
Another influential model is that of Alexander and Crutcher (1990), who proposed four parallel circuits linking different parts of the cerebral cortex with specific basal ganglia and thalamic nuclei. Each circuit mediates different functions. For example, the ‘limbic’ circuit, which is involved in emotional and motivational processes, links the anterior cingulate cortex and medial prefrontal cortex with the ventral striatum, ventral pallidum, and mediodorsal thalamus.
Thalamus, basal ganglia, and cerebellum
A variety of cognitive and psychiatric consequences have been described following lesions of subcortical nuclei. These structures, which were previously considered to be primarily involved in sensory processing (the thalamus) or in motor control (the basal ganglia and cerebellum) are now all known also to be integrally involved in cognition and behaviour, and lesions therein may present with psychiatric as well as neurological features (DeLong and Wichmann, 2007; Schmahmann and Pandya, 2008), and include disturbances of memory, language, and mood. Reduced initiation of actions is also characteristic of basal ganglia pathology, and impaired consciousness with thalamic lesions.
Rostral brainstem
Behavioural disturbances frequently accompany lesions of the rostral brainstem. The most characteristic features are an amnestic syndrome (see below), hypersomnia, and the syndrome of akinetic mutism (‘vigilant coma’) or stupor.
White matter
As well as the corpus callosum, mentioned earlier, damage to other white matter tracts—both subcortical and periventricular—has important neuropsychiatric and behavioural consequences. For example, degeneration of the white matter (leukodystrophy) can produce a schizophrenia-like syndrome (Hyde et al., 1992), whereas multiple focal areas of white matter damage are associated with an increased risk of mood disorder and dementia. The clinical features of white matter pathology depend on the location of the damage and whether it is focal or diffuse. For a review of this subject, see Schmahmann et al. (2008).
Memory systems and their neuroanatomy
Clinical, neuropsychological, and brain imaging studies (both structural and functional) support the existence of multiple memory systems in the human brain. These functions may all be affected more or less selectively by brain lesions. The most basic division lies between implicit (i.e. procedural) and explicit (i.e. declarative) memory. The former includes a range of phenomena that are not usually subject to conscious analysis, such as motor skills, conditioned behaviours, and repetition priming. Explicit memory is subclassified into episodic (memory of autobiographical events) and semantic (knowledge of the world) functions.
The short-term store underpins working memory (e.g. when dialling an unfamiliar telephone number). Distinct anatomical substrates for short-term storage of verbal and visuospatial information, both controlled by a central executive, have been proposed. In neuropsychological terms, ‘short-term’ refers to immediate recall. By contrast, the concept of ‘short-term’ memory, as sometimes applied by clinicians to recall over minutes and days, does not correspond to an anatomical substrate.
Specific types of memories, such as faces and topographical information, may engage dedicated subsystems. Episodic memory has both anterograde (new learning) and retrograde (recall of past events) components. It appears to be mediated by a network of cortical and subcortical structures, which include the hippocampus, parahippocampal and entorhinal cortices, amygdala, mammillary bodies, fornix, cingulate, thalamus, and frontobasal cortex, whereas semantic memory may be subserved by a partly independent network overlapping the language areas. Broadly speaking, verbal memories are mediated by the left (dominant) hemisphere and non-verbal memories by the right hemisphere.
For a review of the classification and neuroanatomical basis of memory and its dysfunction, see Budson and Price (2005) and David and Kopelman (2009).
Assessment of the ‘neuropsychiatric patient’
The assessment of cognitive function was introduced in Chapters 1 and 3 as part of the general psychiatric assessment, and in this chapter the evaluation of amnesia, delirium, and dementia will be discussed. In this section, we introduce key aspects of the initial approach to the patient who has a suspected neuropsychiatric disorder. For a more detailed discussion, see Kipps and Hodges (2005) and David (2009b).
Before embarking upon the assessment, it is worth bearing in mind the range of major diagnostic possibilities. One question to be addressed early on is whether there is clouding of consciousness, as this defines delirium, and the assessment can proceed to determine its cause. If there is no impaired consciousness, the main diagnostic categories to consider are amnesia, dementia, or a ‘functional’ cause of cognitive impairment. The key feature of an amnestic syndrome is a specific deficit in episodic memory, as outlined above; although rare, amnestic syndrome needs to be considered in patients presenting with memory impairment, especially in those with alcohol dependence. A functional cause should always be considered, as memory impairment may occur secondary to many psychiatric disorders; in particular, depression in the elderly may present as pseudodementia (see Chapter 18, p. 503). Distinction between organic and functional causes requires positive evidence to be sought for both forms of disorder, and is an important distinction to make, as it has a significant impact upon treatment and prognosis. Once these other causes of cognitive impairment (delirium, amnestic syndrome, and functional disorder) have been ruled out, a provisional diagnosis of dementia can be made (assuming that the impairment is of sufficient severity), and attention can then turn to determining the type of dementia from which the patient is suffering, as discussed later.
History and mental state examination
Although physical examination and laboratory investigations play a much larger role than elsewhere in psychiatry, the history remains essential: ‘the difference between a good neuropsychiatrist and a mediocre one is a good history’ (David, 2009b). An informant is especially important, as the presence of impaired cognition or consciousness will necessarily limit the patient’s ability to provide a full and accurate history. Key points in the history include the onset, duration, and progression of the impairment—for example, an acute onset suggests delirium or, if it began after a fall, may indicate a subdural haematoma. The neurological, medical, and family history is important too, as many causes of cognitive impairment are secondary to pre-existing disorders, or have a genetic basis.
Physical examination
The physical examination needs to be comprehensive and careful, as signs may not be conspicuous. Particular attention should be paid to the nervous system, as well as to searching for peripheral stigmata of systemic disease and alcohol dependence. Specific signs may provide diagnostic clues (Cooper and Greene, 2005) (e.g. the Argyll–Robertson pupil of neurosyphilis, optic disc pallor in vitamin B12 deficiency, or cranial nerve signs in neurosarcoidosis).
Investigations
The choice and extent of investigations will depend on the findings from the history, mental state examination, and physical examination, but usually includes a core set of tests, such as the cognitive tests and blood tests used in evaluation of dementias noted below. In difficult or atypical cases, and in younger patients, investigations may be extensive, and the opinion and assistance of a neurologist, physician, or neurosurgeon may be required.
Some examples of specialized investigations used in neuropsychiatric evaluation are listed below.
• Structural brain imaging with CT or MRI. Neuroimaging can detect focal and diffuse pathologies, and longitudinal scans can map progressive changes that mirror clinical decline. MRI is superior to CT for most purposes, including evaluation of white matter disease, and the ability to perform volumetric measurements. Functional brain imaging with fMRI, MRS, SPECT, or PET is a valuable research tool, but is not in widespread clinical use.
• Neuropsychological testing is less widely used than previously, in part because of the increasing availability of brain imaging. However, it can still play a valuable part in characterization of the cognitive impairment (e.g. the cognitive domains that are most affected), inferred localization of the lesion (see p. 313), and measurement of severity and progression (David, 2009b). Rating scales for the assessment of dementia are considered below.
• Electroencephalogram (EEG) studies retain a limited but valuable role in several situations where EEG findings are characteristic—for example, in delirium, prion disease, and detection of non-convulsive status epilepticus. They are also useful in the differential diagnosis of stupor, as a normal EEG would suggest a dissociative state.
• Cerebrospinal fluid (CSF) examination after lumbar puncture is essential if an inflammatory or infective process is suspected. It may also become more widely used in the evaluation of dementia, as different proteins are being found which have diagnostic or prognostic value.
• Genetic testing has a key role in the diagnosis (and prediction) of a very limited range of disorders in which the mode of inheritance, and the causative gene, are known (e.g. Huntington’s chorea).
• Brain biopsy, usually of the right frontal lobe, is occasionally indicated as a last resort in the diagnosis of unexplained cognitive impairment or in suspected prion disease. However, the risks of this procedure must always be weighed against the diagnostic and prognostic information that will be obtained from it.
Delirium
Delirium is characterized by global impairment of consciousness (clouding of consciousness), resulting in reduced level of alertness, attention, and perception of the environment. A number of other terms, such as ‘confusional state’ and ‘acute organic syndrome’, have also been used, but delirium is the preferred term in both ICD-10 and DSM-IV.
For a review of delirium and its management, see Burns et al. (2004) and Meagher and Trzepacz (2009).
Epidemiology
Delirium occurs in 15–40% of patients in general medical or surgical wards, and a higher proportion of patients in intensive-care units (Siddiqi et al., 2006). It is more common in the elderly and in other individuals with diminished ‘cerebral reserve’, notably those with preexisting dementia. Overall, about one in five general hospital inpatients develops delirium at some stage of their admission.
Clinical features
The cardinal feature is disturbed consciousness. It is manifested as drowsiness, decreased awareness of one’s surroundings, disorientation in time and place, and distractibility. At its most severe the patient may be unresponsive (stuporose), but more commonly the impaired consciousness is quite subtle. Indeed the first clue to the presence of delirium is often one of its other features, which include mental slowness, distractibility, perceptual anomalies, and disorganization of the sleep–wake cycle (see Table 13.2).
Symptoms and signs vary widely between patients (Meagher et al., 2007), and in the same patient at different times of day, typically being worse at night. For example, some patients are hyperactive, restless, irritable, and have psychotic symptoms, while others are hypoactive, with retardation and perseveration. Repetitive, purposeless movements are common in both forms. Thinking is slow and muddled, but often rich in content (‘dream-like’). Ideas of reference and delusions (often persecutory) are common, but are usually transient and poorly elaborated. Visual perception is often distorted, with illusions, misinterpretations, and visual hallucinations, sometimes with fantastic content. Tactile and auditory hallucinations also occur. Anxiety, depression, and emotional lability are common. The patient may be frightened, or perplexed. Experiences of depersonalization and derealization are sometimes described. Attention and registration are particularly impaired, and on recovery there is usually amnesia for the period of the delirium.
Aetiology
The main causes of delirium are listed in Table 13.3. Often more than one cause contributes. Old age, frailty, and previous medical and neurological disorders lower the threshold for developing delirium.
Table 13.2 Clinical features of delirium
Impaired attention |
Disorientation for time and place |
Impaired memory |
Psychotic-like symptoms |
Perceptual disturbances |
Delusions |
Perplexity |
Thought disorder |
Behavioural and other symptoms |
Agitation |
Irritability |
Labile affect |
Word-finding difficulties |
Temporal course |
Rapid onset |
Fluctuation over 24-hour period |
Reversal of sleep–wake cycle |
The pathophysiological basis of delirium is unclear. The severity of clinical disturbance correlates with the degree of slowing of cerebral rhythms on EEG, and the neurotransmitters dopamine and acetylcholine are implicated in a final common pathway.
Management of delirium
General principles
Delirium is a medical emergency. It is essential to identify and treat the underlying cause, and a range of investigations may be required (see Table 13.4). Diagnostic instruments, such as the Confusion Assessment Method, can also be valuable (Wong et al., 2010). As delirium is often caused by drugs (due to side-effects or withdrawal effects), these should always be suspected until there is evidence of another cause. As well as urgent investigations, general measures are necessary to relieve distress, control agitation, and prevent exhaustion. These include frequent explanation, reorientation, and reassurance. Unnecessary changes in the staff who are caring for the patient should be avoided. The patient should ideally be nursed in a quiet single room. Relatives should be encouraged to visit regularly. At night, lighting should be sufficient to promote orientation, while not preventing sleep. For a review, see Meagher and Trzepacz (2009).
Table 13.3 Causes of delirium
Alcohol intoxication Alcohol withdrawal and delirium tremens Opiates Prescribed drugs Any drug with anticholinergic properties Any sedative Digoxin Diuretics Lithium Steroids |
Medical conditions Febrile illness (e.g. urinary tract infection) Septicaemia Organ failure (cardiac, renal, hepatic) Hypo- or hyperglycaemia Post-operative hypoxia Thiamine deficiency |
Neurological conditions Epileptic seizure (post-ictal) Head injury Space-occupying lesion Encephalitis Cerebral haemorrhage |
Other Constipation Dehydration Pain Sensory deprivation |
Drug treatment
Drug treatment of the underlying physical problem should be reviewed to ensure that it is the minimum required. Despite the above interventions, which should always be tried first, many patients with delirium require medication to control agitation and distress, and to allow adequate sleep.
There is a lack of good-quality trials of delirium treatment (Skrobik, 2010). In practice, the drug of choice is usually an antipsychotic. Haloperidol is conventionally used, in a dose carefully titrated to achieve the desired calming effect without excess sedation or side-effects. If necessary, the first dose can be given intramuscularly, followed by doses every 6 hours (typically 2–10 mg per day, although elderly patients may require less). Atypical antipsychotics are also increasingly used instead of haloperidol. Some causes of delirium require avoidance of antipsychotics, or particular caution when using them. This includes all patients with coexisting dementia, especially dementia with Lewy bodies, who are particularly sensitive to antipsychotics.
Table 13.4 Investigations for delirium
Full blood count Urea and electrolytes Renal function tests Liver function tests Calcium Random blood glucose Blood cultures Arterial blood gas Syphilis serology |
Other tests Urinalysis Chest X-ray Drug screen Cardiac enzymes MRI or CT brain scan EEG |
Antipsychotics should be avoided in delirium associated with alcohol withdrawal (delirium tremens, see p. 450) or with epilepsy, because of the risk of seizures. In delirium tremens, a reducing regimen of the benzodiazepine chlordiazepoxide is the standard treatment (see p. 459).
All sedative drugs should be used sparingly in liver failure because of the danger of precipitating hepatic coma.
Outcome
Many cases recover rapidly. The prognosis is related to the underlying cause, and is worse in the elderly, and in those with pre-existing dementia or physical illness. There is an elevated mortality rate following delirium, with an estimated 25% mortality at 3 months, although published estimates vary markedly (Siddiqi et al., 2006). A recent meta-analysis of elderly patients found that an episode of delirium was robustly associated with a twofold increased risk of death in the next 2 years, as well as with increased risks of institutionalization and of a diagnosis of dementia (Witlox et al., 2010).
Amnesia and amnestic syndromes
Amnesia is loss of memory, and amnestic syndromes or amnestic disorders are those in which memory is specifically and persistently affected (see Table 13.5). An amnestic disorder is defined by DSM-IV as a specific impairment of episodic memory, manifested as inability to learn new information (anterograde amnesia) and to recall past events (retrograde amnesia), accompanied by ‘significant impairment in social or occupational functioning’, and with evidence of a general medical condition ‘aetiologically related to the memory impairment.’ Unlike dementia, the memory deficit occurs in the absence of evidence for generalized intellectual dysfunction. Korsakov syndrome (also called Korsakoff syndrome) is sometimes erroneously referred to synonymously with amnestic syndrome, but is in fact a specific form of it, as described below.
For a review of this subject, see Kopelman (2009).
Table 13.5 Causes of amnesia
Transient global amnesia Transient epileptic amnesia Head injury Alcoholic blackouts Post-electroconvulsive therapy Post-traumatic stress disorder Psychogenic fugue Amnesia for criminal offence |
Persistent (amnestic syndrome) Korsakov syndrome Herpes encephalitis Posterior cerebral artery and thalamic strokes Head injury |
Clinical features
The cardinal feature is a profound deficit in episodic memory. The full clinical picture is striking. There is disorientation for time, loss of autobiographical information (often extending back for many years), severe anterograde amnesia for verbal and visual material, and lack of insight into the amnesia. Events are recalled immediately after they occur, but forgotten a few minutes later. Thus the digit span, which tests the short-term memory store, is typically normal. New learning is grossly defective, but retrograde memory is variably preserved and shows a temporal gradient, with older memories being better preserved. Other cognitive functions are relatively intact, although some emotional blunting and inertia are often observed.
The other classic feature, seen particularly in Korsakov syndrome, is confabulation, in which gaps in memory are filled by a vivid and detailed but wholly fictitious account of recent activities which the patient believes to be true. The confabulating patient is often highly suggestible.
Aetiology and pathology
Amnesia results from lesions in the medial thalamus, other midline diencephalic structures, or medial temporal lobes (hippocampus and adjacent temporal cortex). Cases due to damage in the medial temporal lobe typically produce the ‘purest’ amnesia, with little in the way of disorientation or confabulation; these features are characteristic of thalamic and diencephalic lesions.
Korsakov syndrome
The commonest cause of amnestic syndrome is Korsakov syndrome, named after the Russian neuropsychiatrist (sometimes spelt as Korsakoff) who described it in 1889. The alternative term, Wernicke–Korsakov syndrome, was proposed by Victor et al. (1971), because the syndrome often follows an acute neurological syndrome called Wernicke’s encephalopathy, described by Wernicke in 1881, consisting of delirium, ataxia, pupillary abnormalities, ophthalmoplegia, nystagmus, and a peripheral neuropathy. Korsakov syndrome is usually caused by thiamine deficiency, secondary to alcohol abuse, although it occasionally results from hyperemesis gravidarum and severe malnutrition. The classic neuropathological findings are neuronal loss, gliosis and microhaemorrhages in the periaqueductal and paraventricular grey matter, the mammillary bodies, and the anterior and mediodorsal thalamus. Other causes of amnestic syndrome include tumours and infarcts in the medial thalamus (diencephalic amnesia), and encephalitis (see below).
For a review of Korsakov syndrome, see Kopelman et al. (2009).
Investigation and management
Alertness to the possibility of amnestic syndrome is essential; the patient may not fit the stereotype of chronic alcohol misuse associated with Korskov syndrome, and this is a potentially reversible condition. Useful findings from investigations include a reduced red cell transketolase level, which is a marker of thiamine deficiency, and an increased MRI signal in midline structures.
In practice, Korsakov syndrome should be assumed to be the cause of amnestic syndrome until another aetiology (see Table 13.6) can be demonstrated, and should be treated urgently with thiamine without awaiting the results of investigations. Thiamine is given parenterally in an acute presentation, together with rehydration, general nutritional support, and treatment of supervening alcohol withdrawal. Thiamine replacement should always precede administration of intravenous glucose-containing solutions. Close liaison with physicians and neurologists is important.
Table 13.6 Differential diagnosis of transient amnesia and other paroxysmal neuropsychiatric symptoms
Syncope (cardiogenic, vasovagal, reflex) Transient ischaemic attacks Migraine Epileptic seizure (ictal or post-ictal) Hypoglycaemia Phaeochromocytoma Transient global amnesia Narcolepsy and other parasomnias Tonic spasms of multiple sclerosis Treatment-related complications in Parkinson’s disease Drug abuse Medial temporal lobe tumour |
Functional Panic attacks and hyperventilation Dissociative disorder Schizophrenia Bipolar affective disorder Aggressive outburst in personality disorder Temper tantrums (in children) Breath-holding spells (in children) |
In the longer term, persistent amnestic syndrome may require substantial rehabilitation and support, as the condition markedly impairs normal activities and ability to provide self-care.
Course and prognosis
In the series of Victor et al. (1971), consisting of 245 patients with Wernicke–Korsakov syndrome, 96% of the patients presented with Wernicke’s encephalopathy. Mortality was 17% in the acute stage, and 84% of the survivors developed a typical amnestic syndrome. There was no improvement in 50% of cases, complete recovery in 25%, and partial recovery in the remainder. Favourable prognostic factors were a short history before diagnosis and prompt commencement of thiamine replacement.
The prognosis is poor in cases of amnestic syndrome due to encephalitis and other causes of irreversible bilateral hippocampal or diencephalic damage. However, amnestic syndrome due to head injury has a better outlook. Progressive amnesia suggests a slowly expanding structural lesion, such as a midbrain tumour.
Transient global amnesia
The syndrome of transient global amnesia is important in the differential diagnosis of paroxysmal neurological and psychiatric disturbance (see Table 13.6). It occurs in middle or late life. The clinical picture is of sudden onset of isolated, often profound, anterograde amnesia in a clear sensorium, generally lasting for between 15 minutes and 24 hours. Functional imaging studies during transient global amnesia have demonstrated localized transient hypo- or hyperperfusion consistent with dysfunction of circuits that mediate episodic memory.
The patient appears bewildered, and requires repeated reorientation, only to ask the same questions moments later. However, there is no disturbance of alertness, and (in contrast to psychogenic fugue) personal identity is retained. Procedural memory is spared—for example, the patient may carry on driving competently during the episode. Apart from the memory disturbance, the neurological examination is entirely normal.
Complete recovery, with amnesia for the period of the episode, is usual and recurrence is rare. However, investigation is always indicated, to exclude other causes of amnesia (see above and Table 13.6). Patients with transient global amnesia often present as emergencies to general practitioners and Accident and Emergency departments, and the syndrome may be misdiagnosed as a dissociative fugue.
For a review of this subject, see Quinette et al. (2006).
Dementia
Dementia is an acquired global impairment of intellect, memory, and personality, but without impairment of consciousness (Burns and Illiffe, 2009a). It is usually but not always progressive. The syndrome of dementia is caused by a range of diseases (see Table 13.7), of which Alzheimer’s disease (accounting for 50–60% of cases), vascular dementia (20–25%), and dementia with Lewy bodies (15–20%) are the commonest. Only a small proportion (4% of cases in one large series) are currently potentially reversible (Hejl et al., 2002).
Although dementia is a global or generalized disorder, it often begins with focal cognitive or behavioural disturbances. However, both DSM-IV and ICD-10 definitions require impairment in two or more cognitive domains (memory, language, abstract thinking and judgement, praxis, visuoperceptual skills, personality, and social conduct), sufficient to interfere with social or occupational functioning. Deficits may be too mild or circumscribed to fulfil this definition, and are then called mild cognitive impairment (see p. 325).
In this section, the main features of the dementia syndrome are described, followed by the principles of assessment. We then discuss the clinical, aetiological, and neuropathological features of the major diseases that produce dementia. Note in Table 13.7 that many other neuropsychiatric disorders and some medical disorders can also include cognitive impairment; these conditions are considered later in this chapter and in Chapter 15, respectively. Dementia as a result of substance misuse, especially alcohol misuse, is discussed in Chapter 17.
It should be noted that the management of dementia, and the relationships between dementia and ageing, are deferred until Chapter 18.
Clinical features of dementia
The presenting complaint is usually of poor memory. Other features include disturbances of behaviour, language, personality, mood, or perception.
Primary neurodegenerative disorders Alzheimer’s disease*, dementia with Lewy bodies, Pick’s disease and other frontotemporal dementias,* Parkinson’s disease*, prion diseases*, Huntington’s disease* |
Vascular causes Vascular dementia, multiple strokes, focal thalamic and basal ganglia strokes, subdural haematoma |
Inflammatory and autoimmune causes Systemic lupus erythematosus and other vasculitides with CNS involvement, Behçet’s disease, neurosarcoidosis, Hashimoto’s encephalopathy, multiple sclerosis |
Trauma Severe head injury, repeated head trauma (‘dementia pugilistica’) |
Infections and related conditions HIV, iatrogenic and variant CJD (prion disease), neurosyphilis, post-encephalitic |
Metabolic and endocrine causes Renal failure, hepatic failure, hypothyroidism, hyperthyroidism (‘apathetic’ or masked), hypoglycaemia, Cushing’s syndrome, hypopituitarism, adrenal insufficiency |
Neoplastic causes Intracranial space-occupying lesions, carcinomatous or lymphomatous meningitis, paraneoplastic limbic encephalitis |
Post radiation Acute and subacute radionecrosis, radiation thromboangiopathy |
Post anoxia Severe anaemia, post-surgical (especially cardiac bypass), carbon monoxide poisoning, cardiac arrest, chronic respiratory failure |
Vitamin and other nutritional deficiencies Vitamin B12 deficiency, folate deficiency |
Toxins Alcohol, poisoning with heavy metals, organic solvents, organophosphates |
Other Normal-pressure hydrocephalus Leucodystrophy* |
* Causes exist in genetically determined forms.
The clinical picture is much determined by the patient’s premorbid personality. People with good social skills may continue to function adequately despite severe intellectual deterioration. Dementia is often exposed by a change in social circumstances or an intercurrent illness. The elderly, socially isolated, or deaf are less likely to compensate for failing intellectual abilities; however, their difficulties may go unrecognized or be dismissed.
Forgetfulness is usually early and prominent, but may sometimes be difficult to detect in the early stages. Impaired attention and concentration are common and non-specific. Difficulty with new learning is usually the most conspicuous feature. Memory loss is more evident for recent than for more remote material. Disturbed episodic memory is manifested as forgetfulness for recent day-to-day events, with relative preservation of procedural memory (e.g. how to ride a bicycle) and, at least initially, general knowledge about the world at large. By contrast, words and, ultimately, the very objects to which they refer, lose their meaning for patients with semantic memory impairment (as in certain frontotemporal dementias).
Loss of flexibility and adaptability in new situations, with the appearance of rigid and stereotyped routines (‘organic orderliness’), and, when taxed beyond restricted abilities, sudden explosions of rage or grief (‘catastrophic reaction’) are frequent. As dementia worsens, patients are less able to care for themselves and they neglect social conventions. Disorientation for time, and later for place and person, is common. Behaviour becomes aimless, and stereotypies and mannerisms may appear. Thinking slows and becomes impoverished in content and perseverative. False ideas, often of a persecutory kind, gain ground easily. In the later stages, thinking becomes grossly fragmented and incoherent. This is reflected in the patient’s speech, with syntactical and dysnomic errors. Eventually the patient may become mute. Mortality is increased, with death often following bronchopneumonia and a terminal coma (Mitchell et al., 2009).
Behavioural, affective, and psychotic features often accompany the cognitive deficits during dementia. They appear to be part of the underlying biology of the disease process, although in the early stages, while insight is retained, they may also be a psychological response to the realization of cognitive decline. Mood disturbances are particularly common, together with distress, anxiety, irritability, and sometimes aggression. Later, emotional responses become blunted, and sudden, apparently random mood changes occur. Psychotic symptoms are also a common and fluctuating feature during dementia.
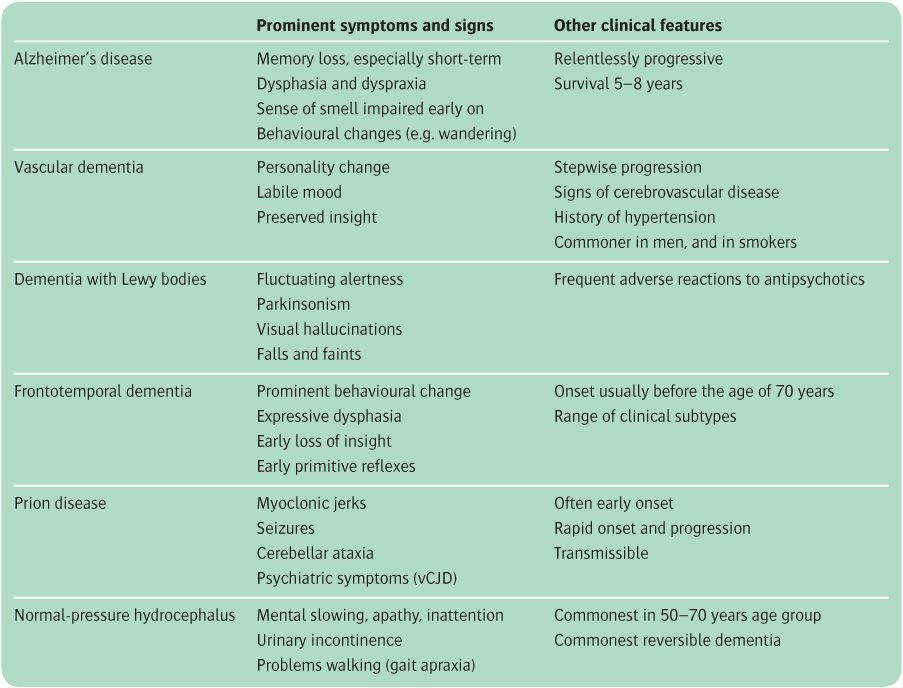
The balance of these core symptoms and signs, together with some additional features, forms the basis for the clinical differentiation between the various causes of dementia, as summarized in Table 13.8 and described in the following sections.
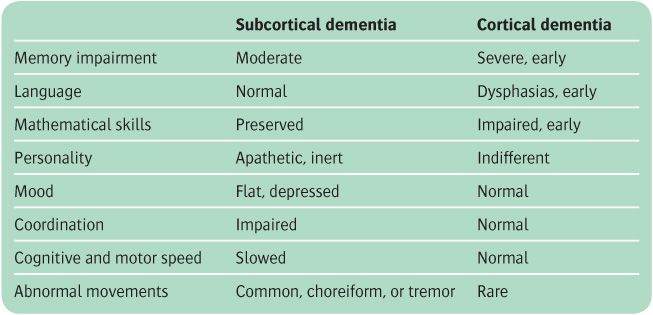
Subcortical and cortical dementia
A distinction is sometimes drawn between subcortical and cortical dementia, based upon their putative neuro-anatomical basis. Although the distinction is blurred, clinically and pathologically, the terms have descriptive utility (Turner et al., 2002). The key features are summarized in Table 13.9, and examples of the diseases are listed in Table 13.10. The term subcortical dementia is seen to refer to a syndrome of slowness of thought, difficulty with complex, sequential intellectual tasks, and impoverishment of affect and personality, with relative preservation of language, calculation, and learning. It contrasts with the spectrum of dysfunction (including early, prominent impairments of memory, word finding, or visuospatial abilities) that is seen in cortical dementias.
Table 13.8 Clinical features that help to distinguish between major causes of dementia
Presenile and senile dementia
Another traditional distinction was that made between dementia occurring in those under 65 years of age (prese-nile or earlyonset dementia) and dementia beginning later in life (senile or late-onset dementia). It arose in part because of the belief that the major causes were different—Alzheimer’s disease in the former, and vascular dementia in the latter. With the realization that Alzheimer’s disease is the commonest form of dementia in both groups, less attention is now paid to this categorization. However, presenile dementia does differ in certain respects from later-onset dementia (Sampson et al., 2004). Frontotemporal dementia and prion disease are relatively more common, vascular dementia is rarer, and a higher proportion of cases are due to genetic diseases. These factors influence management. Patients with presenile dementia are more likely to be referred to and investigated by neurologists, before psychiatrists become involved in their care. Investigations are more intensive and extensive because of the importance of making a definite diagnosis and providing a clear prognosis. The course tends to be more rapid.
Table 13.9 Features of cortical and subcortical dementias
Assessment of dementia
Assessment of a patient who is presenting with a complaint of cognitive impairment involves several stages. A key question to be addressed initially is whether the impairment is due to dementia. This involves ruling out other causes, notably delirium, amnesia, and depression. Other patients will have mild cognitive impairment.
Having established the probable diagnosis of dementia, its characteristics (including severity, symptom and behaviour profile, and associated risks) are considered, together with assessment of its cause, and identification of potentially reversible causes. In the study mentioned earlier, the latter consisted mostly of hydrocephalus and space-occupying lesions (Hejl et al., 2002).
Table 13.10 Examples of cortical, subcortical, and mixed causes of dementia
Alzheimer’s disease Frontotemporal dementias |
Subcortical Huntington’s disease Parkinson’s disease Focal thalamic and basal ganglia lesions Multiple sclerosis |
Vascular dementia Dementia with Lewy bodies Corticobasal degeneration Neurosyphilis |
Assessment of the severity and clinical profile
Screening tests are useful in the assessment of dementia and its severity, and for monitoring progression (Salmon and Bondi, 2009). Different scales are available to assess cognition, behavioural symptoms, global functioning and activities of daily living, and depression; the latter is useful because depression can coexist with dementia and worsen functioning. Some commonly used screening tests are listed in Table 13.11. For cognitive impairment, the benchmark is the Mini-Mental State Examination (MMSE), which has a sensitivity and specificity for dementia of around 80% when a cut-off score of 23 is chosen (Mitchell, 2009). The ‘seven-minute screen’ and clock test are valuable and brief alternatives. The ADAS-Cog is specifically designed for suspected Alzheimer’s disease, and is widely used in clinical trials to monitor treatment response. The CAMCOG is largely a research tool.
Careful evaluation of the behavioural symptoms of dementia is an integral part of the assessment, and needs to be repeated during the illness, since these symptoms are common and they pose as many difficulties for carers as the cognitive symptoms.
Assessment of the cause of dementia
Definitive diagnosis of the cause of dementia can usually only be made neuropathologically or, in rare cases, by identification of genetic mutations. However, the differing profiles of the various dementias allow ‘probable’ diagnoses to be made by experienced clinicians with reasonable accuracy. For example, Burns et al. (1990) found that 88% of cases of clinically diagnosed Alzheimer’s disease were confirmed at autopsy. The use of biochemical, radiological, and genetic investigations only modestly increases the diagnostic accuracy for common dementias, but is important for ruling out rarer and reversible causes. With regard to the latter, Heijl et al. (2002) found that about 4% of patients with dementia who were referred to a memory clinic had a potentially reversible form. Table 13.12 summarizes the investigations for dementia. An MRI or CT scan is recommended as routine in many guidelines, but in practice brain scanning, like most investigations, is used in some but not all patients. The extent to which investigations are carried out, as well as the addition of more specialized tests, depends upon the patient’s age and history, the results of the initial tests, and the subsequent differential diagnosis.
Table 13.11 Screening tests for dementia
Mini-Mental State Examination (MMSE) Six-Item Cognitive Impairment Test Seven-minute screen Clock drawing test Hopkins Verbal Learning Test (HVLT) Mental Test Score (MTS) Alzheimer’s Disease Assessment Scale—cognitive subscale (ADAS-cog) Cambridge Examination for Mental Disorders of the Elderly, cognitive section (CAMCOG) |
Behavioural and psychological features Neuropsychiatric Inventory MOUSEPAD BEHAVE-AD Cohen–Mansfield Aggression Inventory |
Activities of daily living Bristol Scale Alzheimer’s Disease Functional Assessment and Change Scale Disability Assessment for Dementia |
Depression Cornell Scale Geriatric Depression Rating Scale |
Global assessment Clinical Dementia Rating (CDR) |
Table 13.12 Investigations for establishing the cause of dementia
Full blood count Erythrocyte sedimentation rate Urea and electrolytes Liver function tests Calcium and phosphate Thyroid function tests Vitamin B12 and folate |
In secondary care MRI or CT brain scan Urinalysis Syphilis serology HIV status Chest radiograph Neuropsychological assessment Genetic testing EEG |
Adapted from Burns and Illiffe (2009a).
Assessment of risk in dementia
Patients with dementia are at risk from self-neglect, poor judgement, wandering, and abuse. Their physical health is often a problem. Risks to others may occur because of aggressive or disinhibited behaviour. Fitness to drive is a specific issue to consider, and may be difficult to determine in the early stages (Breen et al., 2007); current UK regulations allow those with ‘sufficient skills’ and whose ‘progression [of dementia] is slow’ to continue to drive, subject to annual review. Thus risk assessment is part of a full assessment of dementia. A good history from carers and other informants is essential, and an occupational therapist has an important role to play in assessment of functional ability.
Early detection of dementia
Mild cognitive impairment
Increasing attention is being given to the earlier diagnosis of dementia in those with equivocal evidence, or subjective complaints, of worsening memory, in part driven by the research focus on developing treatments to delay or prevent progression to dementia. This intermediate category, which was introduced in ICD-10, is called mild cognitive impairment (MCI). However, its clinical significance remains unclear, as only 5–10% of patients convert to dementia each year, and the majority of MCI patients have not progressed to dementia 10 years later (Mitchell and Shiri-Feshki, 2009). The MMSE is of limited value in the MCI population (Mitchell, 2009). Neuropathologically, MCI is mainly associated with Alzheimer-type changes, and also with vascular pathology. For a review of MCI, see Petersen et al. (2009).
Presymptomatic diagnosis and biomarkers
There is increasing evidence that dementia, especially Alzheimer’s disease, can be detected premorbidly, long before overt symptoms of any kind are present. Longitudinal studies show that there are selective and characteristic neuropsychological impairments, detectable up to 20 years before the onset of symptoms, as well as functional brain changes (e.g. in regional glucose metabolism) and structural and neuropathological abnormalities that precede symptoms by several years. For example, hippocampal atrophy at the rate of about 2% per annum begins several years before the symptoms of Alzheimer’s disease appear. This contrasts with normal age-related decreases of 0.5% per year (Frisoni et al., 2010).
Brain imaging that is used in this way is an example of a biomarker—that is, a test or marker which is of value in diagnosis, prognosis, or prediction of the treatment response. A range of biomarkers are nearing clinical utility in Alzheimer’s disease and some other dementias, including measurements of proteins in the CSF and blood (Blennow et al., 2010), and PET detection of β-amyloid deposits in the brain (Jagust et al., 2009).
Alzheimer’s disease
In 1907, Alois Alzheimer reported the case of Auguste D, a woman with presenile dementia whose brain exhibited unusual neuropathological features. It was Alzheimer’s colleague, Emil Kraepelin, who named the disease (Maurer et al., 1997). For many years the disease was thought to be rare and limited to presenile forms of dementia, but classic studies by Roth and colleagues (Blessed et al., 1968) suggested that it is the commonest cause of senile dementia, a conclusion which has been confirmed by many subsequent studies. About 60% of dementia is attributable to Alzheimer’s disease (Neuropathology Group of the Medical Research Council Cognitive Function and Ageing Study, 2001).
Prevalence rates for populations over 65 years of age are 2–7% for moderately or severely affected individuals. Age-specific prevalence rates approximately double with every additional 5 years of age, from about 1% at 65 years, rising to around 8–10% at age 80 years and 30–40% at age 90 years (Nussbaum and Ellis, 2003). Because of the ageing population, the projected numbers of cases, and their health costs, will increase substantially (Mebane-Sims, 2009).
For a general review of Alzheimer’s disease, see Burns and Illiffe (2009b) and Lovestone (2009a).
Clinical features
The main features of dementia have been described above, and Table 13.13 summarizes the clinical features of Alzheimer’s disease (strictly, ‘dementia of the Alzheimer type’, since formal diagnosis awaits neuropathology) (see also Table 13.8, and Lovestone, 2009a).
The first evidence of the condition is often minor forgetfulness which may be difficult to distinguish from normal ageing. The condition progresses gradually for the first 2–4 years, with increasing memory disturbance and lack of spontaneity. Memory is lost for recent events first. Language is usually affected early on, with difficulty in finding words or naming objects, and impairments in the ability to construct fluent and informative sentences. Visuospatial skills may be affected, with difficulties in tasks such as copying pictures or learning the way round unfamiliar environments (e.g. when on holiday or in an unfamiliar house). Disorientation in time gives rise to poorly kept appointments and changes in the diurnal pattern of activity.
Table 13.13 Key clinical features of Alzheimer’s disease
Memory impairment (amnesia), with gradual onset and continuing decline Aphasia Apraxia Agnosia Disturbance in executive functioning (e.g. planning, reasoning) |
Other features Depression Psychosis Behavioural symptoms (e.g. agitation, wandering) Personality change |
Depression
The relationship between Alzheimer’s disease and depression is complex. Depression is a probable risk factor for the disease, may be confused with it, or may occur as part of the syndrome. Regarding the latter point, major depression occurs in about 10% of cases, with less marked episodes and symptoms occurring in over 50% of cases. Patients who experienced depression have greater decreases in serotonin and noradrenergic markers than other patients with Alzheimer’s disease.
Psychotic symptoms
Delusions and hallucinations occur in a significant minority of patients at some stage in the illness. Their prevalence is unclear, as many studies did not distinguish Alzheimer’s disease from dementia with Lewy bodies (see below). Recent estimates suggest rates of 10–50% for delusions and 10–25% for hallucinations. The commonest delusions are persecutory, concerning theft (however, as this idea often arises from the patient’s forgetfulness, it is questionable whether it is helpful to regard this as a true delusion).
Behaviour
Changes in behaviour are common, and are of particular concern to carers. The patient may be restless and wake at night, disorientated and perplexed. Motor activity may increase in the evening (‘sundowning’), and eventually the sleep–wake cycle may become completely disorganized. Aggression (both verbal and physical) is common, and often takes the form of resistance to help with personal care. Serious physical violence towards others is rare. Both increases and reductions in level of activity are common, involving varying degrees of purposefulness. Wandering can refer to a variety of different behaviours, but patients may place themselves at risk by going into unsafe environments. Patients with dementia may under- or overeat, with associated changes in weight and nutritional state. Changes in sexual behaviour occur, usually with a reduction in drive, although sexual disinhibition occasionally occurs.
Self-care and social behaviour decline, although some patients maintain a good social facade despite severe cognitive impairment, particularly if carers are able to assist with these functions.
Course
In the early stages of Alzheimer’s disease, the clinical features are modified by the patient’s premorbid personality, and their traits tend to be exaggerated. In the middle and later stages of the illness, the cognitive impairments increasingly predominate, together with the neurological and behavioural features noted above. Incidental physical illness may cause a superimposed delirium, resulting in a sudden deterioration in cognitive function. Median survival from diagnosis is 5–7 years, and is slightly less in men than in women. Shorter survival is also associated with an older age of onset, and a rapid rate of cognitive decline.
Investigations
The investigation of suspected Alzheimer’s disease follows the same principles as the investigation of dementia in general, outlined above. In addition, a comment on genetic testing is warranted. Such testing is indicated in the very rare cases of familial early-onset Alzheimer’s disease for which three causative genes are known (see below). Although the apoE4 allele of the apolipoprotein E gene is a major risk factor in all forms of Alzheimer’s disease (see Box 13.1 and also see below), routine testing for this variant in the differential diagnosis of dementia is not currently recommended, and there is no place for genetic screening of healthy subjects in order to predict future dementia. The scientific and ethical issues involved have been discussed by Green et al. (2009).
Neuropathology
On gross examination the brain is shrunken, with widened sulci and enlarged ventricles. Brain weight is reduced. On microscopic examination, the cardinal diagnostic features are neurofibrillary tangles and senile plaques (also called amyloid plaques) in the cerebral cortex and many subcortical regions. The diagnostic criteria are based upon the abundance and distribution of plaques and tangles—the Consortium to Establish a Registry for Alzheimer’s Disease (CERAD) criteria (Mirra et al., 1991). However, it should be noted that these features are also seen, to some extent, in some non-demented elderly people (e.g. Savva et al., 2009), and the relationship between clinical dementia and neuropathology is more complex than is sometimes assumed to be the case.
In addition to the tangles and plaques, there is selective loss of neurons in the hippocampus and entorhinal cortex, proliferation of astrocytes (gliosis), and loss of synapses. The latter is the strongest neuropathological correlate of cognitive impairment. Other histological findings include amyloid deposits in blood vessel walls (so-called vascular amyloid or congophilic angiopathy), Hirano bodies (intracellular, crystalline deposits) and granulovacuolar accumulation (vacuoles or ‘holes’ within neurons).
For a review of the neuropathology of Alzheimer’s disease, see Duyckaerts et al. (2009).
Progression of neuropathology
The disease starts in the entorhinal cortex, before spreading to the hippocampus, association areas of the parietal lobe, and some subcortical nuclei. Six neuropathological stages based upon β-amyloid deposition are recognized, called Braak stages, which correlate with clinical severity (Braak and Braak, 1991). The spread of pathology along cortico-cortical projections leads to an effective ‘disconnection’ between affected regions.
Senile (amyloid) plaques
Senile plaques are deposits of insoluble proteins, together with degenerating neurites (neuronal processes) and glia. They occur in the space between neurons (the neuropil). Both neuritic and diffuse plaques are recognized, depending on their appearance using silver stains; neuritic plaques have a dense-staining core, whereas diffuse plaques have been likened to cotton wool. The neuritic plaques are pathologically more significant. The protein at the heart of all senile plaques is β-amyloid (also called Aβ or A4), a 39–42 amino-acid peptide. This molecule and its encoding gene are central to the aetiology of the disease (see below).
Neurofibrillary tangles
Neurofibrillary tangles occur within the cell body of neurons, especially pyramidal neurons of the cerebral cortex and hippocampus. They are formed of paired helical filaments, which in turn are comprised of the microtubule-associated protein tau. The normal function of tau is in axonal transport and maintenance of the neuronal cytoskeleton. Tangles are thought to occur because tau becomes hyperphosphorylated, rendering it insoluble. The presence of a tangle causes dysfunction and death of the neuron.
Aetiology and pathogenesis
Genes
In rare families, usually those with an early onset of illness (before the age of 60 years), an autosomal-dominant mode of inheritance can be discerned. Causative mutations have been identified in three genes—amyloid precursor protein (APP, on chromosome 21), presenilin 1 (PS1, on chromosome 14), and presenilin 2 (PS2, on chromosome 1). Discovery of APP as the first ‘Alzheimer gene’ was a seminal event in psychiatry (see Box 13.1). These three genes together account for the majority of familial cases of the disease; equally, at least one other gene is likely to exist. Different mutations are known in each gene, and the age of onset, features, and progression of disease vary depending on the causative mutation.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


