HISTORICAL PERSPECTIVE
The primary objective of this chapter is to provide an updated review on the diagnosis, classification, etiology, and pathophysiology of dystonia. Therapeutic approaches are covered separately (see Chapter 30). One of the earliest descriptions of dystonia was provided by Gowers in 1888, who coined the term “tetanoid chorea” to describe the movement disorder in two siblings later found to have Wilson’s disease (1,2) (Table 28.1). The term “dystonia musculorum deformans,” coined by Oppenheim in 1911, was criticized because fluctuating muscle tone was not necessarily characteristic of the disorder; the term “musculorum” incorrectly implied that the involuntary movement was due to a muscle disorder, and not all patients became deformed. Until recently, the term “torsion dystonia” has been used in the literature, but since torsion is part of the definition of dystonia, the term “torsion dystonia” seems redundant and, hence, the simple term “dystonia” is preferred.
PHENOMENOLOGY AND DIFFERENTIAL DIAGNOSIS OF DYSTONIA
Dystonia is currently defined as a neurologic syndrome characterized by involuntary, sustained, patterned, and often repetitive muscle contractions of opposing muscles, causing twisting movements or abnormal postures. Traditional descriptions of dystonia emphasize that the muscle contractions are sustained and that rapid dystonic movements are often not recognized. These rapid movements resemble myoclonus, and the term “myoclonic dystonia” is sometimes applied when the movements are fast and repetitive. Myoclonic dystonia, a rapid dystonic movement, should be differentiated from the syndrome of myoclonus–dystonia, an autosomal dominant, alcohol-responsive disorder manifested by coexisting dystonia and myoclonus, where the myoclonus occurs in body parts not necessarily affected by dystonia. This definition has been recently revised by a consensus committee as follows: “Dystonia is defined as a movement disorder characterized by sustained or intermittent muscle contractions causing abnormal, often repetitive, movements, postures, or both. Dystonic movements are typically patterned and twisting, and may be tremulous. Dystonia is often initiated or worsened by voluntary action and associated with overflow muscle activation and “mirror” dystonia, triggered by volitions repetitive movement in the contralateral, unaffected, limb (3,4). The consensus proposed that dystonia is classified along two axes: clinical characteristics, including age at onset, body distribution, temporal pattern, and associated features (additional movement disorders or neurologic features); and etiology, which includes nervous system pathology and inheritance (3).
A characteristic feature of dystonia that helps to distinguish it from the other hyperkinetic movement disorders is the fact that dystonic movements, whether slow or rapid, are repetitive and patterned. The term “patterned” refers to the repeated involvement of the same group of muscles. Thus, a patient with cervical dystonia manifested by torticollis to the left tends to always have the same abnormal pattern and direction of movement (i.e., turning of the head to the left) during the course of the disease. Although additional muscles may subsequently be involved, the pattern usually remains the same. This is in contrast to chorea, which consists of brief movements that occur continuously and flow randomly from one body part to another.
Tics are brief and intermittent movements (motor tics) or sounds (phonic tics) (5). In contrast to dystonia, tics can be more easily suppressed, are usually abrupt rather than continual, and are often preceded by a subjective, compulsive urge or sensation (premonitory sensation) that is temporarily relieved after the tic has been executed. The phenomenology of tics ranges from brief, lightning-like jerks (clonic tics) to more sustained contractions (tonic or dystonic tics). Although these latter tics resemble dystonic movements, they can be differentiated easily from dystonic movements, particularly when clonic tics or other features of Tourette’s syndrome are present.
The diagnosis of dystonia is often complicated by the coexistence of tremor. Two basic types of tremor can occur in patients with dystonia: postural (essential-like) and dystonic. Postural tremor in the hands, phenomenologically identical to essential tremor (ET), is present in one-quarter of patients with cervical dystonia (6). Whether the hand tremor seen in about 25% of patients with cervical dystonia represents an enhanced physiologic tremor, ET, dystonic tremor, or some other form of postural tremor is unknown. Although patients referred to a movement disorders clinic have disorders more severe and atypical than the general population of patients with ET-like tremor, the true prevalence of dystonia in patients with ET tremor is clearly higher than in the general population. Furthermore, a number of large families have been described in which some members have only ET, while others have only dystonia and still others have the combination of dystonia and ET (7). Münchau et al. (8) studied 11 patients with classic ET and compared them to 19 patients with cervical dystonia and arm tremor. They found that the latency of second agonist burst during ballistic wrist flexion movements occurred later in ET patients than in those with arm tremor–associated cervical dystonia. Furthermore, the latter group had a greater variability in reciprocal inhibition than the ET group. Patients with normal presynaptic inhibition had onset of their arm tremor simultaneously with their cervical dystonia (mean age 40 years), whereas patients with reduced or absent presynaptic inhibition had onset at an earlier age (mean 14 years) and the interval between the onset of the tremor and of cervical dystonia was longer (mean 21 years). These findings suggest that the mechanisms of arm tremor in patients with ET and cervical dystonia are different. In some patients, head and trunk tremor (usually of slow, 2- to 5-Hz frequency) may precede the onset of dystonia and may be the initial manifestation of focal dystonia (dystonic tremor). Some patients with dystonic tremor present with asymmetric rest-hand tremor and decreased arm swing, which may lead to initial misdiagnosis of Parkinson’s disease (PD) (9). Certain task-specific (e.g., writing) tremors may actually represent forms of focal dystonia (10). Rarely, cerebellar and parkinsonian tremors can be associated with dystonia, and dystonia may be the presenting finding of PD or other parkinsonian disorders as progressive supranuclear palsy (PSP) and corticobasal degeneration (11,12).
| Milestones in the History of Dystonia |
Year | Author | Description |
1887 | Wood | Facial and oromandibular dystonia |
1888 | Gowers | Tetanoid chorea—two siblings, later found to have Wilson’s disease |
1901 | Destarac | Torticollis spasmodique—a 17-year-old girl with torticollis, tortipelvis, writer’s cramp, foot cramps, improved by sensory tricks, exacerbated by motor activity |
1903 | Leszynsky | Hysterical spasms and gait |
1908 | Hunt | Myoclonia of the trunk, tic spasms, hystericia |
1908 | Schwalbe | Hereditary tonic, crampus syndrome, maladie des tics—rapid movements; recognized the familial nature of dystonia in Jewish siblings (Lewin family); used scopolamine |
1911 | Ziehen | Torsion neurosis—did not believe it to be hysterical; observed that “convulsive movements increased during voluntary movement” and during emotional excitement |
1911 | Oppenheim | Dystonia musculorum deformans and dysbasia lordotica progressive, “monkey” or “dromedary” gait, “mobile spasms,” sustained posturing, spasms, fluctuating muscle tone, rapid movement resembling tremor, chorea, and athetosis |
1911 | Flatau and Sterling | Progressive torsion spasm—noted hereditary, repetitive pattern, jerky; Jewish patients of high intelligence; objected to the term “deformans” because not all patients become disfigured and objected to the term musculorum because it implied a muscle condition |
1912 | Fraenkel | Rapid, twisting, sustained movement, tortipelvis |
1912 | Wilson | Hepatolenticular degeneration—clonic or tic-like spasms, choreiform, and athetoid movements |
1916 | Hunt | Slow, twisting or clonic, rhythmic movements |
1919 | Mendel | Torison dystonia—review of literature; 33 patients; “a morbid disease entity” |
1920 | Taylor | Dystonia lenticularis, postural (myostatic) and kinetic forms of dystonia |
1926 | Davidenkow | Myoclonic dystonia—rapid tic-like movements |
1929 | Wimmer | “Not a disease but only a syndrome”—seen in Wilson’s disease, postencephalitic, perinatal brain damage |
1944 | Herz | Idiopathic dystonia as a disease entity—15 personal cases and 105 from the literature; “slow, long-sustained, turning movements”; alternating “myorhythmia” or “very rapid, tic-like twitchings” |
1958 | Cooper | Thalamotomy |
1959 | Zeman | Autosomal dominant |
1960 | Zeman et al. | Formes frustes of dystonia |
1962 | Denny-Brown | Fixed or relatively fixed attitude |
1967 | Zeman and Dyken | No specific neuropathology in dystonia brains |
1976 | Marsden | Blepharospasm is a form of focal dystonia |
1983 | Fahn | High-dose anticholinergic therapy |
1983 | Jankovic and Patel | Blepharospasm–oromandibular dystonia secondary to rostral brain stem–diencephalic lesion |
1985 | Scott et al. | Botulinum toxin for blepharospasm |
1989 | Ozeliue et al. | Gene for autosomal dominant dystonia linked to chromosome 9q32–34 (DYT1) |
1989 |
| Botulinum toxin type A approved by the U.S. Food and Drug Administration |
1994 | Ichinose et al. | Mutations in the GTP-cyclohydrolase 1 gene on chromosome 14q22.1–22.2 in autosomal dominant dopa-responsive dystonia |
1995 |
| Mutation in the TH gene on chromosome 11p15.5 causes autosomal recessive form of dopa-responsive dystonia |
1996 |
| Third International Dystonia Symposium |
1997 | Ozelius et al. | Dystonia gene (DYT1) encodes an ATP-binding protein |
1999 | Nygaard et al. | A gene for myoclonus–dystonia mapped to chromosome 7q21–q23 |
1999 |
| Botulinum toxin type A and botulinum toxin type B approved for the management of cervical dystonia by the U.S. Food and Drug Administration |
2001 | Zimrich et al. | Myoclonic dystonia localized to 7q21 gene coding for ε-sarcoglycan |
2005 |
| Prospective, controlled study of GPi DBS in dystonia |
Dystonia is either a symptom of an underlying disorder or a specific disease entity (Table 28.2). When no etiologic factor can be identified, the dystonia is referred to as primary dystonia. Primary dystonia can be either sporadic or inherited and is not associated with any cognitive, pyramidal, cerebellar, or sensory abnormalities. When there is an associated neurologic abnormality, such as parkinsonism, dementia, corticospinal tract signs, and other disturbances besides dystonia, the term “dystonia-plus” may be appropriate (see Table 28.2).
CLASSIFICATION OF DYSTONIA
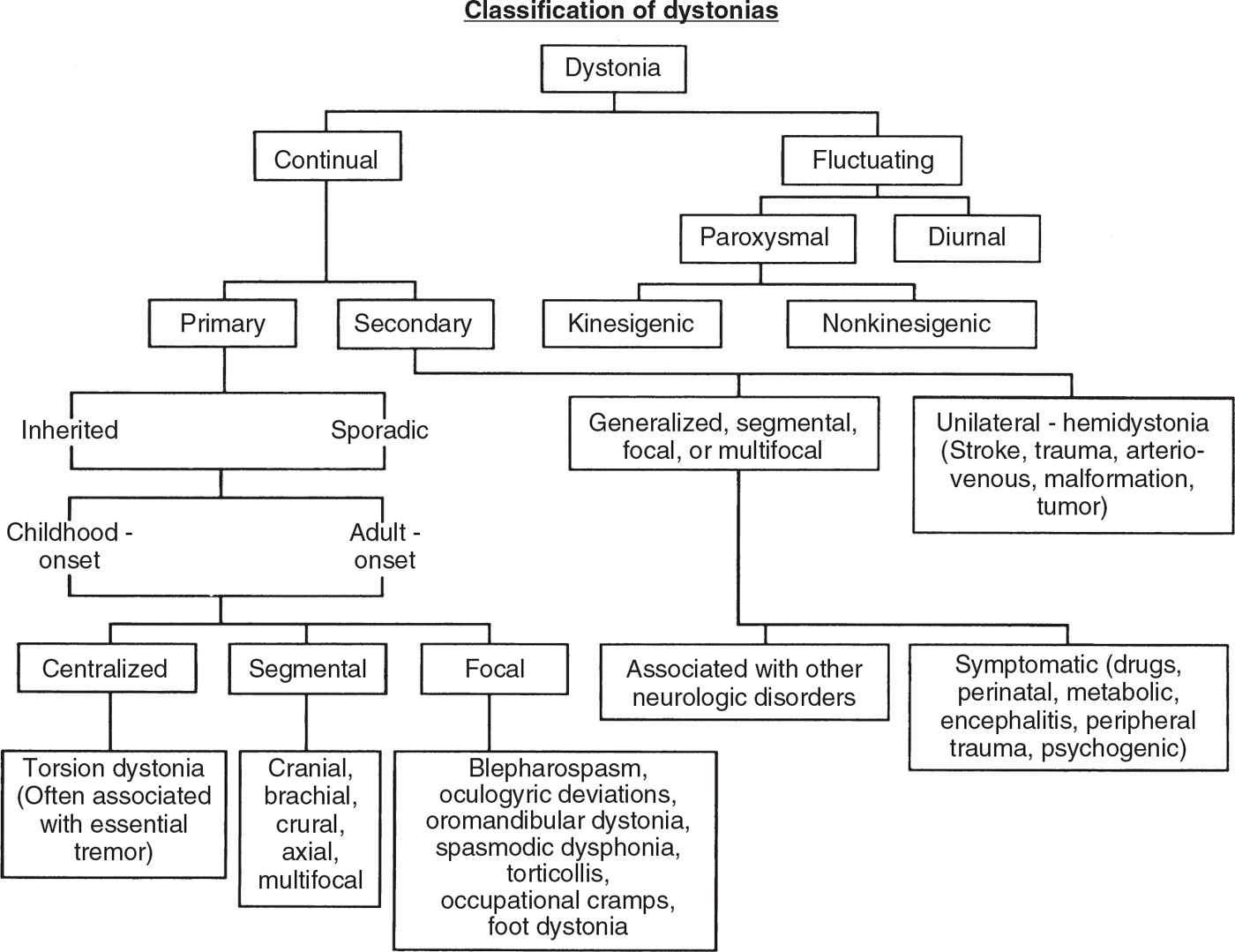
Dystonia can be classified according to (a) severity, (b) clinical characteristics, (c) distribution, (d) age at onset, and (e) etiology (3) (Fig. 28.1).
| Etiologic Classification of Dystonia |
I. Primary dystonia
A. Sporadic
B. Inherited (all autosomal dominant)
Classic (Oppenheim’s) dystonia (common in Ashkenazi Jews, DYT1—9q34)
Childhood- and adult-onset cranial–cervical–limb dystonia (DYT6—8p21–22)
Adult-onset cervical and other focal dystonia (DYT—18p)
II. Secondary dystonia (dystonia-plus syndromes)
A. Sporadic
Parkinson’s disease
Progressive supranuclear palsy
Multiple system atrophy
Corticobasal degeneration
B. Inherited
Autosomal dominant
Dopa-responsive dystonia (DRD) (DYT5—GTP cyclohydrolase I 14q22.1)
Dystonia–myoclonus (11q23)
Alternating hemiplegia of childhood
Dystonia–ataxia (SCA types 3 and 6)
Autosomal recessive
Dopa-responsive dystonia (11p11.5)
Tyrosine hydroxylase deficiency (chromosome–21)
Biopterin deficiency diseases
Aromatic amino acid decarboxylase deficiency (dopamine agonist–responsive dystonia)
III. Heredogenerative diseases (typically not pure dystonia)
A. X-linked recessive
Lubag (X-linked dystonia parkinsonism, DYT3—Xq12–Xq21)
Pelizaeus-Merzbacher disease
Lesch–Nyhan syndrome
Dystonia–deafness (Xq22)
Deafness, dystonia, retardation, blindness
B. Autosomal dominant
Rapid-onset dystonia–parkinsonism
Juvenile parkinsonism–dystonia
Huntington’s disease (IT15–4p 16.3)
Spinocerebellar degenerations (SCA1–SCA8)
Dentatorubropallidoluysian atrophy
Hereditary spastic paraplegia with dystonia
Thalamo–olivary degeneration with Wernicke’s encephalopathy
C. Autosomal recessive
Wilson’s disease (CU-ATPase–13q14.3)
Neurodegeneration with brain iron accumulation type 1 or pantothenate kinase–associated neurodegeneration (formerly Hallervorden–Spatz disease–20p12.3–p13)
Hypoprebetalipoproteinemia, acanthocytosis, retinitis pigmentosa, and pallidal degeneration (HARP syndrome)
Ataxia telangiectasia
Associated with metabolic disorders
1. Amino acid disorders
Glutaric acidemia
Methylmalonic acidemia
Homocystinuria
Hartnup disease
Tyrosinosis
2. Lipid disorders
Metachromatic leukodystrophy
Ceroid lipofuscinosis
Niemann–Pick type C (dystonic lipidosis, “sea blue” histiocytosis)
Gangliosidoses GM1, GM2 variants
Hexosaminidase A and B deficiency
3. Other metabolic disorders
Biopterin deficiency diseases
Triose phosphate isomerase deficiency
Aromatic amino acid decarboxylase deficiency (dopamine agonist–responsive dystonia)
Biotin-responsive basal ganglia disease
D. Mitochondrial
Leigh’s disease
Leber’s disease
E. Unknown inheritance
Neuroacanthocytosis
Rett’s syndrome
Intraneuronal inclusion disease
Infantile bilateral striatal necrosis
Familial basal ganglia calcifications
Hereditary spastic paraplegia with dystonia
Deletion of 18q
Due to a known specific cause
Perinatal cerebral injury and kernicterus: athetoid cerebral palsy, delayed-onset dystonia
Infection: viral encephalitis, encephalitis lethargica, Reye’s syndrome; subacute sclerosing panencephalitis; Creutzfeldt–Jakob disease; HIV infection
Other: tuberculosis, syphilis, acute infectious torticollis
Drugs: levodopa and dopamine agonists, dopamine receptor–blocking drugs, fenfluramine, anticonvulsants, flecainide, ergots, some calcium channel blockers
Toxins: MN, CO, CS2, cyanide, methanol, disulfiram, 3-nitropropionic acid, wasp sting toxin
Metabolic: hypoparathyroidism
Paraneoplastic brainstem encephalitis
Vitamin E deficiency
Primary antiphospholipid syndrome
Cerebrovascular or ischemic injury, Sjögren’s syndrome
Multiple sclerosis
Central pontine myelinolysis
Brain stem lesions
Spinal cord lesions
Syringomyelia
Brain tumor
Arteriovenous malformation
Head trauma and brain surgery (thalamotomy)
Lumbar stenosis
Peripheral trauma (with causalgia)
Electrical injury
IV. Other hyperkinetic syndromes associated with dystonia
A. Tic disorders with dystonic tics
B. Paroxysmal dyskinesias
1. Paroxysmal kinesigenic dyskinesia (16p11.2–q12.1)
2. Paroxysmal nonkinesigenic dyskinesia (2q33–35)
3. Paroxysmal exertional dyskinesia (16p12–q12)
4. Paroxysmal hypnogenic dyskinesia (20q13.2–13.3)
V. Psychogenic
VI. Pseudodystonia
Atlantoaxial subluxation
Syringomyelia
Arnold–Chiari malformation
Trochlear nerve palsy
Vestibular torticollis
Posterior fossa mass
Soft-tissue neck mass
Congenital postural torticollis
Congenital Klippel–Feil syndrome
Isaacs’ syndrome
Sandifier’s syndrome
Satoyoshi’s syndrome
Stiff-man syndrome
Dupuytren’s contractures
Trigger digits
Ventral hernia
CLASSIFICATION BY SEVERITY
Dystonic movements can occur at rest but are usually exacerbated by voluntary motor activity (action dystonia). One form of action dystonia is the task-specific focal dystonia present only during specific activity, such as certain occupations (“occupational cramp,” writer’s cramp) or sports (13). This type of dystonia is exemplified best by writer’s cramp (graphospasm) (14). Other specific activities known to induce dystonic movements or postures include holding a cup or utensils in certain positions, cutting food, typing, and other skilled actions required in certain occupations or sports, including playing musical instruments, such as piano and violin, and use of the lips, tongue, and teeth in embouchure (15,16), auctioneering (17), golfing (18), and other specific activities (19). The severity of dystonia varies from a task- or position-specific dystonia to status dystonicus and dystonic storm, causing a breakdown of the contracting muscles and a life-threatening myoglobinuria (20).
The intensity of dystonic movements can be influenced by various conditions. For example, voluntary motor activity, such as walking, running, writing, talking, and performing specific motor tasks, can intensify dystonia. Furthermore, dystonia often increases with stress and fatigue. On the other hand, dystonic movements sometimes can be relieved by rest, self-hypnosis, and various alleviating maneuvers (AMs), also referred to as sensory tricks, geste antagonistique, or counterpressure, such as touching the chin or the occiput to help overcome torticollis (21). Using electromyography (EMG) to study muscle contractions in patients with cervical dystonia, Schramm et al. (22) proposed a two-phase model in which abnormal head posture is first normalized by counterpressure or volitional antagonistic muscle activity and then the position is stabilized by sensory tricks. Patients with generalized dystonia, particularly involving the trunk and legs, note marked improvement when they walk backward. Rarely, dystonia improves during activity. With this so-called paradoxical dystonia, the patient actively moves the affected body part in an attempt to relive the dystonia. This voluntary movement can be mistaken for restlessness or akathisia, particularly when the dystonia affects the trunk. The type of dystonia that is most typically relieved by voluntary movement, such as speaking, is blepharospasm. Dystonic movements usually cease during sleep but may be recorded during lighter stages of sleep; however, dystonic postures may persist during all stages of sleep.
CLASSIFICATION BY CLINICAL CHARACTERISTICS
Although dystonic movements are usually continual, the timing and intensity of the movements can be influenced by various factors, including emotion, fatigue, relaxation, and motor activity. In some patients, dystonia fluctuates so much that it may be almost absent in the morning and become pronounced and disabling in the afternoons and evenings. This diurnal dystonia may be associated with parkinsonian features and characteristically improves dramatically with levodopa therapy—hence the term “dopa-responsive dystonia” (DRD) (23). The genetics and classification of DRD are discussed later in this chapter.
Another type of fluctuating dystonia is paroxysmal dystonia, characterized by sudden onset or an exacerbation of dystonic movements lasting seconds to hours (24) (also see Chapter 38) (Table 28.3). We favor a classification for this group of disorders that is based chiefly on precipitating events, which categorizes the attacks as either nonkinesigenic or kinesigenic paroxysmal dyskinesia. In contrast to paroxysmal nonkinesigenic dystonia (PND), which occurs unpredictably without any particular precipitant, paroxysmal kinesigenic dystonia (PKD) is induced by a sudden movement. In addition, paroxysmal exertional dystonia (PED) follows prolonged physical exertion, and paroxysmal hypnogenic dystonia (PHD) occurs only during sleep. These paroxysmal episodes usually last only a few seconds to minutes and may be exacerbated by stress, extreme temperatures, menses, and other factors. Some paroxysmal dystonias are also associated with seizures and ataxia. The relationship between paroxysmal dystonia and epilepsy is further supported by the observation that dystonic movements can occur during certain seizures (ictal dystonia) (25) and that PKD responds well to anticonvulsant drugs (24).
In addition to the idiopathic fluctuating dystonias, there are many other causes of paroxysmal or fluctuating dystonias, including seizures, multiple sclerosis, thyrotoxicosis, migraines, transient ischemic attacks, aminoacidurias, and other metabolic disorders (see Table 28.2). Inherited errors of metabolism, such as aromatic amino acid decarboxylase deficiency, should be considered in patients with fluctuating (diurnal) dystonia, particularly if it starts in infancy and if accompanied by axial hypotonia, athetosis, ocular convergence spasm, oculogyric crises, and limb rigidity (26). Patients with aromatic amino acid decarboxylase deficiency fail to respond to levodopa but may obtain substantial benefit from dopamine agonists. The disorder is inherited in an autosomal recessive pattern and the gene maps to 7p12.1–p12.3 (27). Levodopa therapy, acute dystonic reactions to neuroleptics, and gastroesophageal reflux are other examples of intermittent fluctuating dystonia. Another example of intermittent paroxysmal dystonia is oculogyric crisis, characterized by a sudden, transient, conjugate eye deviation, often seen as part of the postencephalitic parkinsonism syndrome, Tourette’s syndrome, drug-induced dystonia, or tardive dyskinesia (28). There are many other causes of paroxysmal dystonias, and these should be pursued particularly in patients with atypical features (24,29).
CLASSIFICATION BY DISTRIBUTION
Dystonia may be classified according to distribution into one of five categories:
1. Focal dystonia affects a single body part and is exemplified by cervical dystonia (torticollis); occupational cramp (e.g., writer’s cramp); foot dystonia; oculogyric deviations; blepharospasm; oromandibular, lingual, pharyngeal, and laryngeal dystonia (spasmodic dysphonia); and some forms of bruxism and trismus.
2. Segmental dystonia affects one or more contiguous body parts. Examples of segmental dystonia include craniocervical dystonia, characterized by the combination of blepharospasm, facial–oromandibular, lingual, pharyngeal, laryngeal, and cervical dystonia. Other categories of segmental dystonia include brachial (one or both arms with or without the involvement of axial or cranial muscles), crural (one leg plus trunk or both legs), and axial (neck and trunk with or without cranial muscles).
3. Multifocal dystonia involves two or more noncontiguous body parts (such as a combination of oromandibular dystonia and dystonia of a leg).
4. Generalized dystonia consists of segmental crural dystonia and dystonia in at least one additional body part (Figs. 28.2 and 28.3).
5. Hemidystonia (unilateral dystonia) involves only half of the body and is usually associated with a structural lesion in the contralateral basal ganglia, particularly the putamen (Figs. 28.4 and 28.5).
Based on a combined series of about 8,000 patients with dystonia evaluated at the Movement Disorders Clinic at Baylor College of Medicine (Houston) and New York-Presbyterian Medical Center (New York), about two-thirds of patients have focal dystonia, one-fourth have segmental dystonia, and one-fourth have either generalized hemidystonia or multifocal dystonia. Cervical dystonia represents about one-half of all patients, cranial dystonia (blepharospasm and oromandibular dystonia) one-fourth, and laryngeal or limb dystonia one-fourth. However, this distribution may not be typical for the general population of dystonic individuals because, by virtue of referral to a specialty clinic, this population is biased toward more severe and atypical cases. It is likely, for example, that dystonic writer’s cramp is much more common in the general population than is indicated by these figures. Several rating scales, including the Toronto Western Spasmodic Torticollis Scale (TWSTRS) (30,31), Barry–Albright Dystonia (BAD) Scale (32), Burke–Fahn–Marsden Scale (33), Unified Dystonia Rating Scale (UDRS), Jankovic Blepharospasm Rating Scale (34), Blepharospasm Disability Scale (35), and Cranial Dystonia Questionnaire (36), are currently used to assess the severity of dystonia.
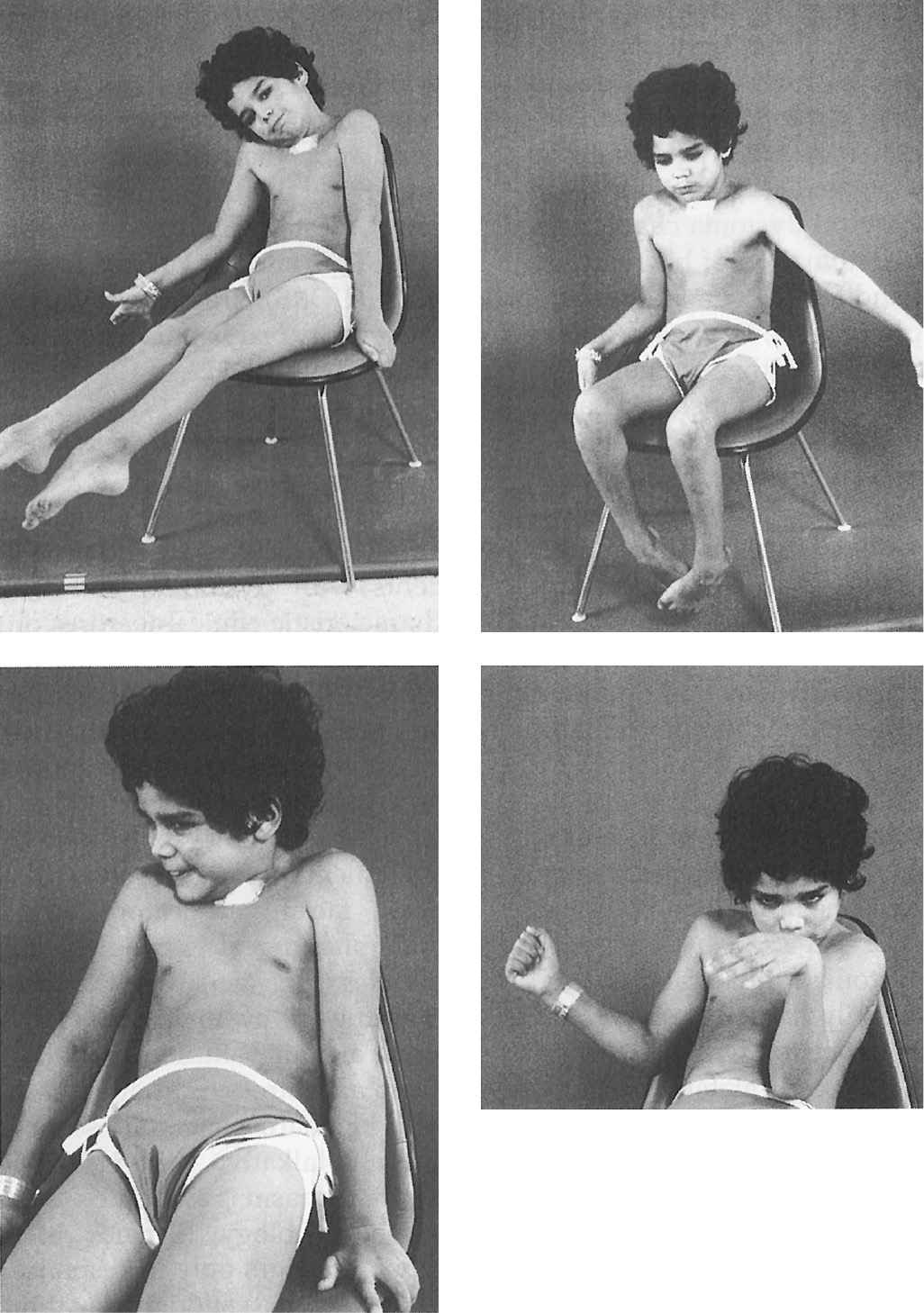
Figure 28.2. An 8-year-old boy with severe inherited generalized dystonia causing myoglobinuria. Tracheostomy was performed because he required prolonged paralysis with curare to prevent further rhabdomyolysis. (Photo courtesy of Joseph Jankovic and Penn, 1982.)
Cranial Dystonia
Since craniocervical structures are most frequently affected by dystonia, the characteristic clinical features of craniocervical dystonia will be emphasized. Blepharospasm, an involuntary bilateral eye closure produced by spasmodic contractions of the entire (pretarsal, preseptal, and periorbital) orbicularis oculi muscles, is often accompanied by dystonic movements of the eyebrows and of the paranasal, facial, masticatory, labial, lingual, oral, pharyngeal, laryngeal, and cervical muscles (Fig. 28.6). Blepharospasm usually affects women (3:1 in comparison to men) older than 50 years (37). Blepharospasm is often exacerbated by exposure to bright light, wind, and air pollution, as well as by movement and stress. Although some patients with blepharospasm may seem somewhat anxious and talkative, there is no evidence that blepharospasm is associated with any overt psychopathology (38). When it occurs only in response to certain stimuli, such as sudden visual threat or auditory or tactile stimuli, the term “reflex blepharospasm” is used. Reflex blepharospasm is also seen in premature infants and patients with various parkinsonian syndromes, nondominant temporoparietal lobe lesions (Fisher’s sign), and a variety of ocular disorders, including blepharitis, conjunctivitis, iritis, and dry-eye syndrome. Eventually, patients have difficulty reading, watching television, driving, and performing other daily activities that depend on normal vision. Various AMs or “sensory tricks,” such as pulling on an upper eyelid, pinching the neck, talking, humming, or singing, can transiently relieve the involuntary eye closure in some patients (39). Because of the fluctuating symptoms, exacerbation by emotional stimuli, and frequent association with logorrhea, anxiety, and depression, there is a tendency to label blepharospasm as a psychogenic problem.

Figure 28.3. A 33-year-old woman with severe, generalized primary dystonia, progressive since age 4.
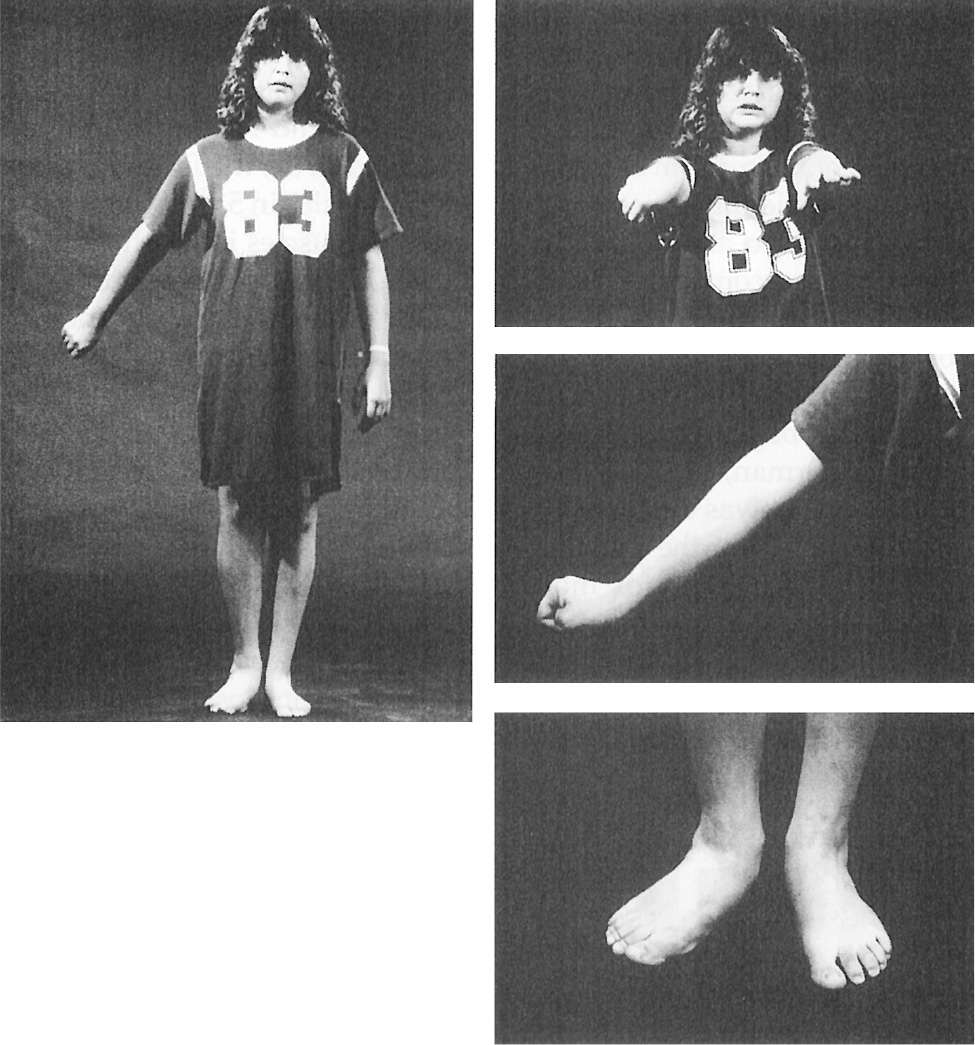
Figure 28.4. A 14-year-old girl with delayed-onset progressive right hemidystonia due to left striatal injury at age 2.
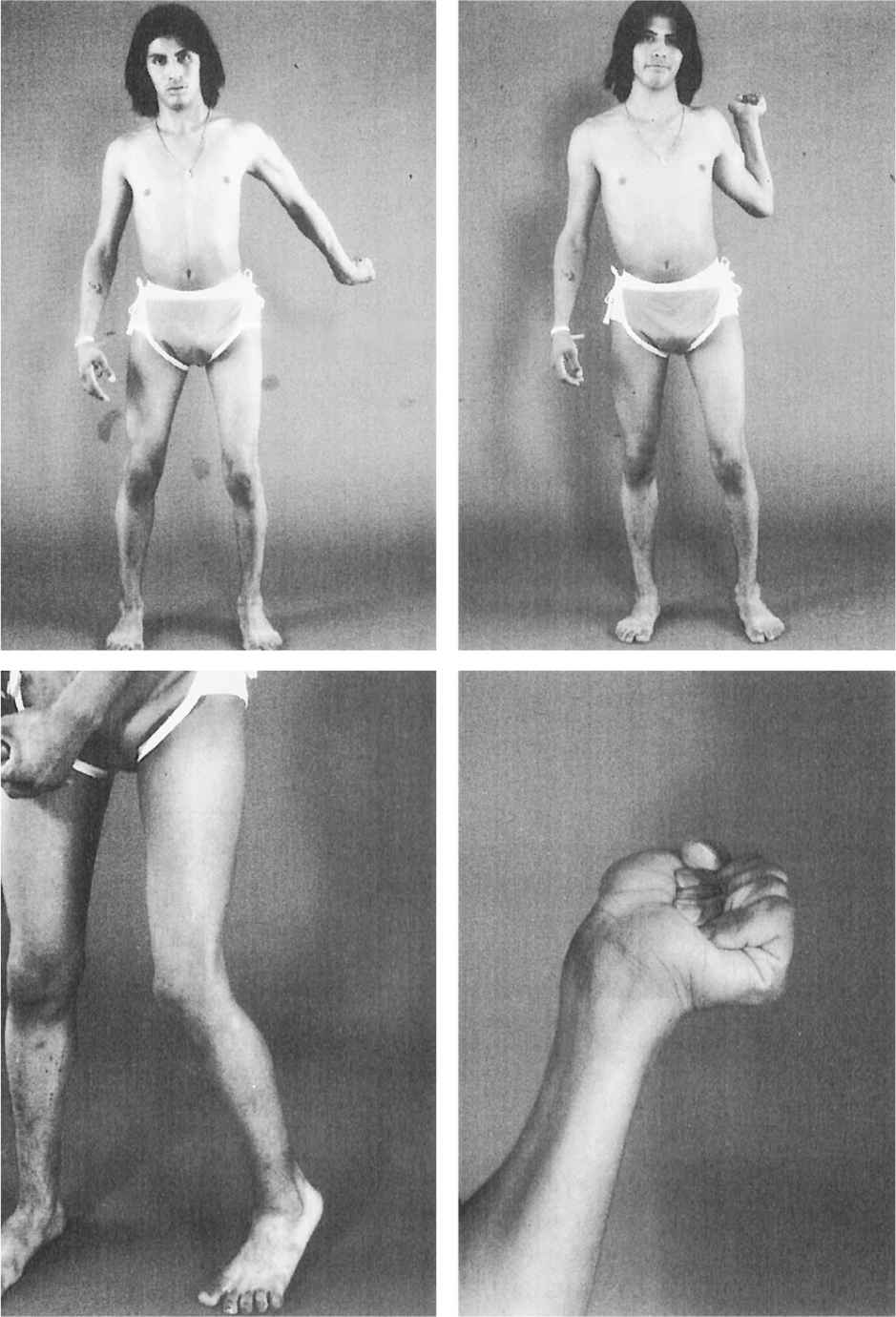
Figure 28.5. Dystonic abduction and extension of the left arm and flexion of the left hand associated with left hemidystonia due to right putamenal postencephalitic lesion.
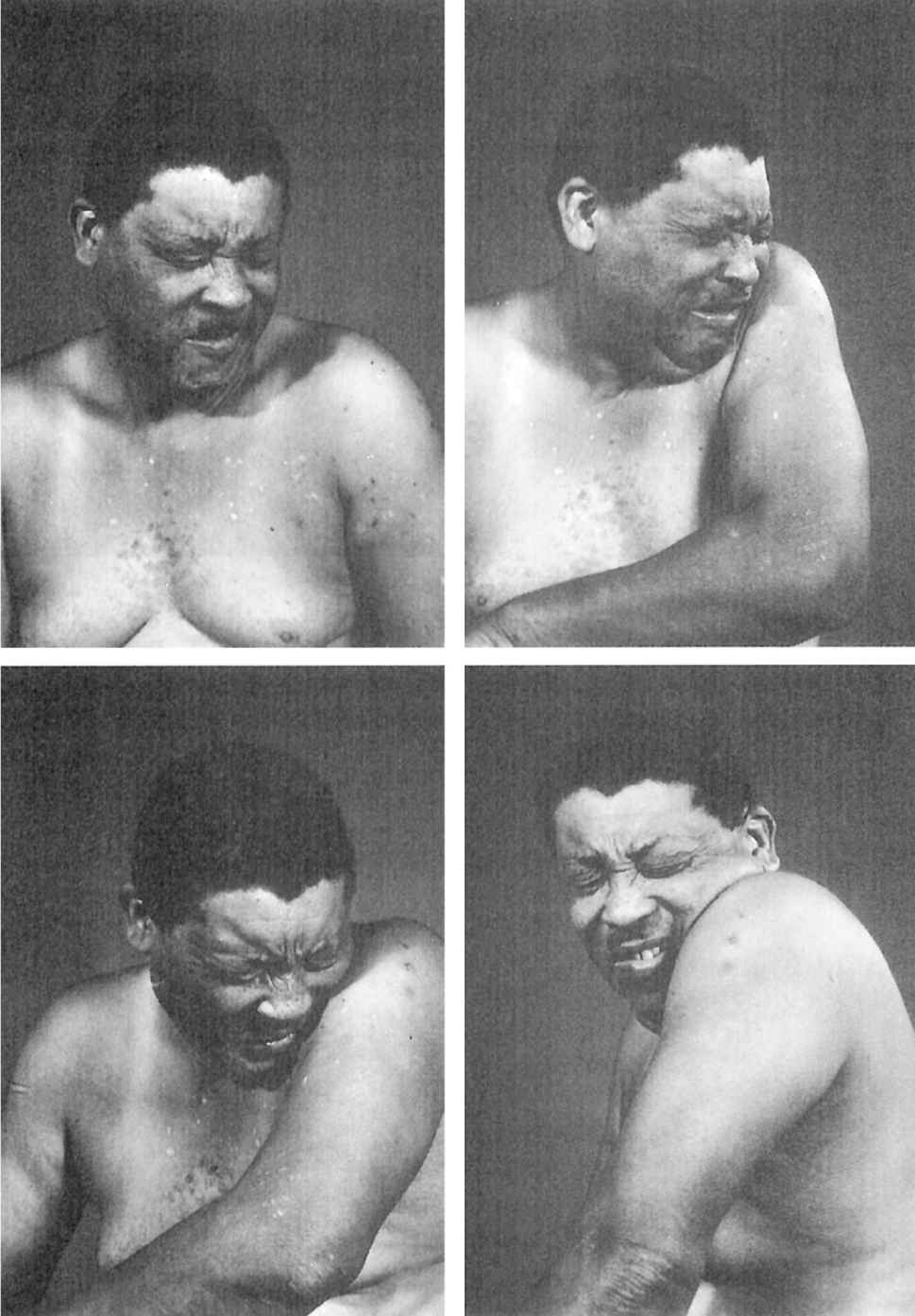
Figure 28.6. Patient with craniocervical dystonia showing blepharospasm, oromandibular dystonia, torticollis, and left shoulder shrug.
A form of focal adult-onset torsion dystonia, blepharospasm can start as an isolated movement of the eyelids. If blepharospasm occurs alone, without the involvement of other craniocervical structures, then the term “essential blepharospasm” might be appropriate. However, in the vast majority of blepharospasm patients, the other facial, pharyngeal, or cervical muscles are also involved, and the term “craniocervical dystonia” best describes the condition. The eponym Meige syndrome, named after the French neurologist who wrote about craniocervical dystonia in 1910, has been used in the literature to describe this movement disorder. However, Horatio Wood, a Philadelphia neurologist, described facial and oromandibular dystonias in 1887, 23 years before Meige. Therefore, the generic term “craniocervical dystonia” is more appropriate for this disorder.
In addition to dystonia, other conditions can lead to closure of the eyelids, such as ptosis due to weakness or paralysis of the levator palpebrae muscle or the smooth muscle of Müller. Some patients are unable to open their eyes because they cannot “activate” the levator palpebrae muscles. This is analogous to the motor blocks or the freezing phenomenon experienced by some, and the terms “apraxia of eyelid opening” and “eyelid freezing” have been used to describe this disorder (37). Apraxia of eyelid opening may occur in isolation without any other motor deficits, or it may be combined with blepharospasm or associated with parkinsonian disorders. The inability to open one’s eyes has been attributed to absence of contraction or even inhibition of the levator palpebrae (despite compensatory frontalis contraction), but others have argued that this sign was caused by isolated contraction of the pretarsal orbicularis oculi. In some cases, EMG recording from the levator palpebrae and orbicularis oculi muscles is required to differentiate this persistent pretarsal orbicularis oculi contraction from the levator inhibition.
Oromandibular dystonia may cause jaw closure with trismus and bruxism, or involuntary jaw opening or deviation, interfering with speaking and chewing and often causing severe pain or discomfort due to secondary temporomandibular joint syndrome (40,41) (Fig. 28.7). Many patients have noted that various maneuvers (sensory tricks) and dental prosthetic devices relieve their jaw spasms, particularly jaw closure dystonia (42). Oromandibular dystonia may follow jaw injury or surgery and may be complicated by secondary dental wear and temporomandibular joint syndrome (43). Besides primary or peripherally induced dystonia, oromandibular dystonia may be associated with or secondary to a variety of neurodegenerative disorders, such as neuroacanthocytosis (44), Huntington’s disease (44), and brainstem lesions (45). Oromandibular dystonia should be differentiated from hemifacial or hemimasticatory spasm, tetany, tetanus, trismus, and mechanical disorder of the jaw or the temporomandibular joint (40,46).
Blepharospasm is usually idiopathic, but many cases are associated with lesions in the rostral brain stem or the basal ganglia (47). A variety of lesions in this area, including stroke, multiple sclerosis, encephalitis, thalamotomy, hydrocephalus, and autoimmune and other disorders, can produce craniocervical dystonia. An abnormal excitatory drive from the basal ganglia to the facial and other motor brain stem nuclei has been proposed as a mechanism of the blepharospasm seen after lesions in the midbrain–diencephalic area (48). Some support for this hypothesis is provided by neurophysiologic studies demonstrating increased amplitude and duration of the R1 and R2 blink response and increased duration of the corneal reflex in blepharospasm patients. In addition, acoustic reflex abnormalities have been found in 87% of patients with craniocervical dystonia (49).
As in other forms of dystonia, genetic factors might also be important in craniocervical dystonia. Besides idiopathic or familial forms, some patients with craniocervical dystonia have secondary dystonias (50). These include drug-induced dystonia, such as that seen after withdrawal from dopamine-blocking agents (tardive dystonia) (24,51,52) or caused by levodopa and other drugs known to produce abnormal involuntary movements. Furthermore, an injury to or surgery on the eye, jaw, teeth, and other facial structures can trigger blepharospasm or oromandibular dystonia (43). A corneal lesion has been found to enhance the gain of blink reflex and as such has been proposed as an animal model for dystonic blepharospasm (53). Postmortem studies of brains of patients with craniocervical dystonia usually show no specific pathology. Some postmortem studies of brains of patients with primary adult-onset dystonia have demonstrated a significant increase in copper levels and a reduction of copper-transporting Menkes protein in the lentiform nuclei (as well as reduced Menkes mRNA copies and lower copper levels in leukocytes) compared with controls (54).
Cervical Dystonia
Cervical dystonia is the most common form of focal dystonia encountered in a movement disorders clinic (55). In addition to turning (torticollis), flexing (anterocollis), or extending (retrocollis) of the neck, the head might be shifted forward or off the midline to either side or tilted toward one shoulder (Fig. 28.8). Frequently, the shoulder is elevated and displaced anteriorly on the side toward which the chin is pointing. In 300 patients with cervical dystonia studied at Baylor College of Medicine (Houston), 61% of whom were women, the mean age was 49.7 years, and the mean duration of dystonia was 7.8 years (56). Torticollis was present in 82%, laterocollis in 42%, retrocollis in 29%, and anterocollis in 25%. A majority (66%) of the patients had a combination of these abnormal postures; in addition, scoliosis was present in 39%. In addition to cervical involvement, 16% of patients had oral dystonia, 12% mandibular dystonia, 10% hand/arm dystonia, and 10% blepharospasm.

Figure 28.7. Patient with oromandibular jaw-opening dystonia that interferes with her ability to eat.
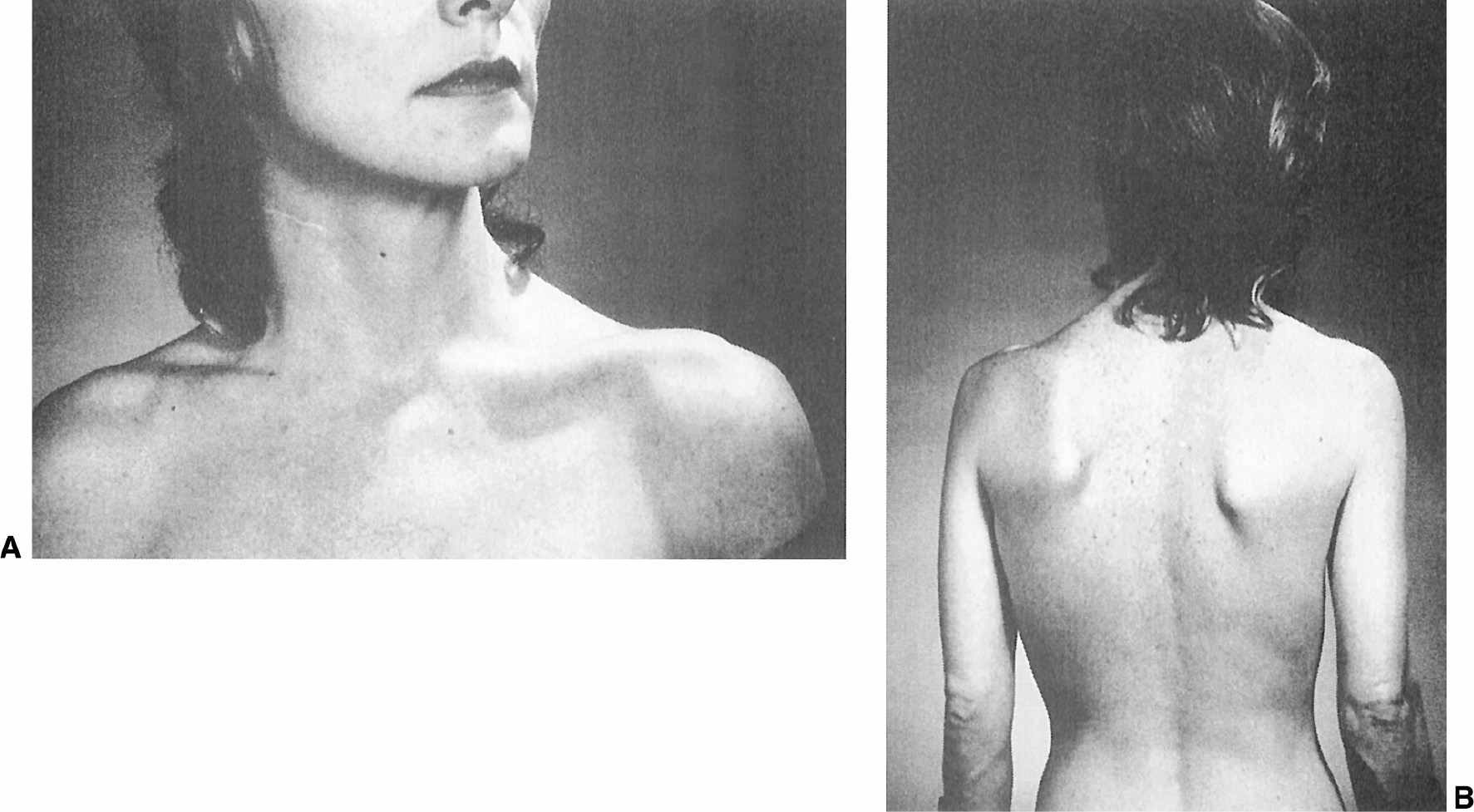
Figure 28.8. A: Patient with torticollis that persists even after right sternocleidomastoid myectomy. B: Same patient with torticollis showing the segmental involvement of the upper trunk and left scapula.
While in some patients the head deviation produced by cervical dystonia is constant, the majority display “spasmodic” contractions of the neck muscles that cause rhythmic, jerky movements of the head in the direction of the most active muscles. These muscles can be identified readily by palpation, which reveals not only the active contractions but also hypertrophy (Fig. 28.9). However, if not performed properly, such an examination can be misleading and may fail to differentiate between the agonist and compensating antagonist muscles, both of which typically contract simultaneously in dystonia. To identify the most involved muscles, we find it helpful to instruct the patient to close his or her eyes and to allow the head to “draw” or deviate into the most comfortable position without any active volitional resistance. EMG recordings of the cervical muscles can be helpful in such an evaluation (57). Blocking the active contraction of the agonist by local injection of an anesthetic or by botulinum toxin (BoNT) causes a reduction in the activity in the antagonist muscle. On the other hand, paralyzing the antagonist muscles should not alter the contraction of the agonist.
Patients with cervical dystonia have been found to have an increased risk of secondary degenerative changes of the upper cervical spine, particularly on the side ipsilateral to the head tilt (58). This cervical arthritis can contribute to the pain, limitation of head movement, and poor response to BoNT and surgical therapy. Evidence of secondary cervical radiculopathy was noted in 32% of the patients at Baylor College of Medicine (56). One patient developed left-arm paralysis due to a thoracic outlet syndrome produced by a hypertrophied left sternocleidomastoid muscle (see Fig. 28.9). The arm weakness improved after a myotomy of the muscle. Torticollis accompanied by pain seems to have a less-favorable prognosis and response to therapy than painless torticollis. Some investigators also have postulated central mechanisms for the pain associated with cervical dystonia (59). In addition to pain, some patients with cervical dystonia also have dysphagia due to delayed swallow initiation (60).
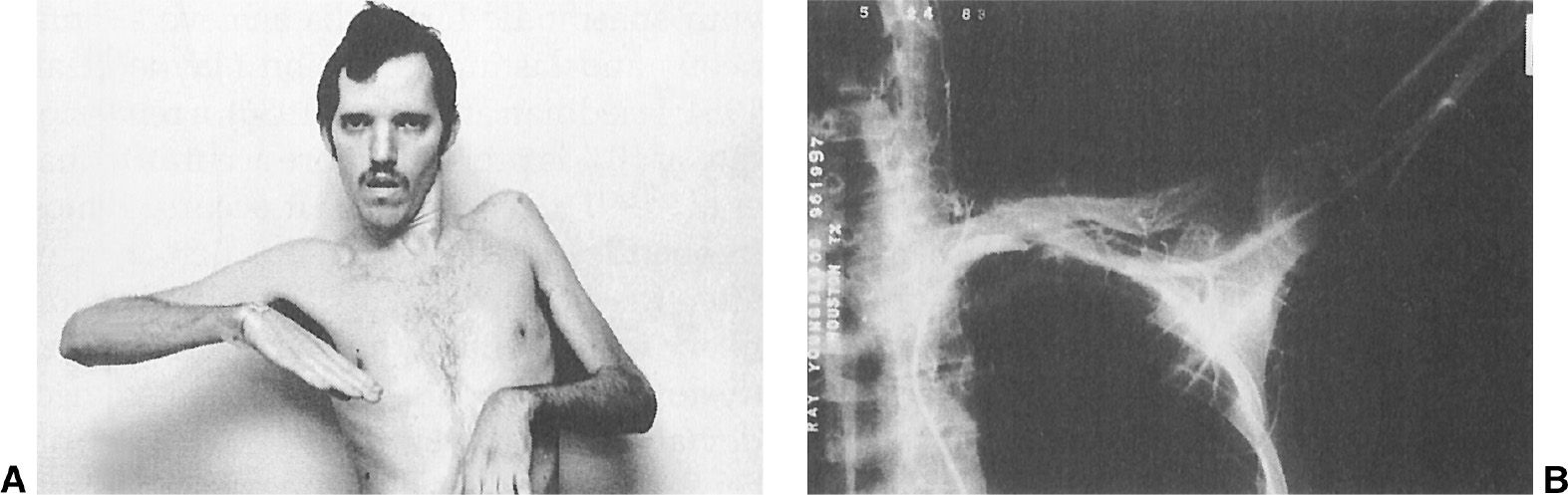
Figure 28.9. A: A 23-year-old schizophrenic man with tardive torticollis and a hypertrophy of the left sternocleidomastoid muscle causing marked left-arm weakness and atrophy due to thoracic outlet syndrome. B: An arteriograph showing occlusion of the left subclavian artery, compressed by enlarged left-neck muscles.
Cervical dystonia is often exacerbated during periods of stress or fatigue and is usually relieved by relaxation and various sensory maneuvers. Although up to quarter of patients with cervical dystonia have been reported to achieve spontaneous and lasting remission, a remission rate of 10% is probably more accurate. Remission, if it occurs, is most frequently noted during the first 3 years after the onset of symptoms and is more likely in patients with spasmodic or “jerky” dystonia than in patients with constant neck deviation. However, in the vast majority of patients, cervical dystonia is a lifelong disorder, and in about 20% of patients, it progresses to segmental or generalized dystonia. Similar to other forms of dystonia, the abnormal muscle contractions that produce head deviation can be temporarily controlled by a variety of sensory tricks, such as touching the chin, face, or back of the head. In a study of 50 patients with cervical dystonia known to have at least one sensory trick, 54% of them had two to five tricks, and 82% had a reduction of head deviation of at least 30% (61). In another study involving 154 patients with cervical dystonia, 138 (89.6%) used some AM, of which 60 (43.4%) reported partial improvement, 55 (39.8%) marked improvement, and 4 (0.03%) no effect on dystonic posture (21). Light touch, usually to the lower face or neck, was utilized by >90%. The presence or location of AM did not correlate with the severity of the dystonia.
Although this observation suggests that cervical dystonia can be influenced by altering the proprioceptive input, the exact mechanism of the counterpressure, sensory trick, or geste antagoniste phenomenon is not known (21,39). In one study of patients with cervical dystonia, using positron emission tomography (PET), Naumann et al. (62) found that keeping the head in primary position by application of a sensory trick decreased motor-cortical activation (including the anterior part of the supplementary motor cortex) contralateral to the side to which the head tends to turn. In addition, the sensory trick is associated with increased activation of the parietal cortex ipsilateral to the direction of dystonic head rotation.
The pathophysiologic mechanisms underlying cervical dystonia have not been elucidated. While some studies have suggested primary disturbance in the vestibular system, other studies concluded that the vestibular hyperactivity noted in some patients with cervical dystonia was secondary (63). Some investigators found that patients with torticollis seemed to relate straight ahead to the orientation of their trunks rather than their heads, thus implying faulty processing of the afferent signals from the vestibular apparatus and from proprioceptors in the neck (39,64). It is possible when torticollis patients utilize a sensory trick that they provide needed additional proprioceptive input to restore their head position. The repetitive head movement seen particularly in phasic cervical dystonia may result in disruption of normal vestibular input and a perception of impaired dynamic equilibrium (65). An involvement of the midbrain in the pathogenesis of cervical dystonia has been suggested by reports of torticollis secondary to midbrain lesions and posterior fossa and spinal cord tumors (66).
Similar to other forms of idiopathic dystonia, genetic mechanisms seem to have an important role in the pathogenesis of cervical dystonia. A family history of some movement disorder was present in 44% (dystonia in 20% and tremor in 32%) of the patients studied at Baylor College of Medicine (56), and a family history of dystonia was present in 12% of the Columbia-Presbyterian patients (67). Tardive dystonia was the cause in 6%, and 11% had onset of their dystonia after a neck trauma. Central and peripheral trauma has been implicated not only in the etiology of some cases of cervical dystonia but also in other forms of focal dystonia (68,69). In contrast to the mobile dystonic posture, seen typically in patients with primary dystonia, the post-traumatic, peripherally induced dystonia is often characterized by a fixed posture, absence of sensory tricks and activation maneuvers, focal muscle hypertrophy, and severe causalgia, sometimes referred to as complex regional pain syndrome (CRPS) (68).
Laryngeal Dystonia
The career of a teacher, a trial attorney, or a professional singer can be prematurely terminated with the development of laryngeal dystonia (spasmodic dysphonia). Despite growing evidence in support of neurologic origin, the symptoms are attributed still too often to psychogenic causes. Dystonia of the larynx may cause excessive and uncontrolled closing of the vocal folds (adductor spasmodic dysphonia), producing effortful and strained voice interrupted by frequent breaks in phonation (voiceless pauses). The abductor form of spasmodic dysphonia is much less common, consisting of prolonged vocal fold openings producing breathy and whispering voice and phonatory pauses extending into vowels. Adductor spasmodic dysphonia is caused by hyperadduction of the thyroarytenoid vocalis complex, and the abductor form of spasmodic dysphonia is due to contractions of the posterior cricoarytenoid muscle. Whereas nearly all cases of adductor spasmodic dysphonia are thought to represent a form of focal dystonia, many cases of abductor dysphonia are thought to be of psychogenic origin. Spasmodic dysphonia often begins as a task-specific focal (laryngeal) dystonia affecting either the speaking voice or the singing voice, but the symptoms usually progress to involve both of them. Many patients with spasmodic dysphonia also have voice tremor, and in some cases, isolated voice tremor precedes by several years the onset of spasmodic dysphonia. Patients with spasmodic dysphonia also may complain of difficulties breathing, and respiratory muscles may be affected in dystonia even without laryngeal involvement (70). Respiratory involvement in dystonia may be manifested by deep inspiratory gasps, loud breathing, respiratory arrests, and respiratory dysregulation.
Limb Dystonia
Idiopathic limb dystonia usually starts as an action dystonia, whereas secondary dystonia may begin as dystonia at rest. The task-specific, focal dystonia seen in many occupational cramps is the most common example of idiopathic arm dystonia. This type of focal dystonia often occurs in association with writing, typing, and feeding; during certain sports-related activities; and during the playing of musical instruments (14,15). Like cervical dystonia, the distal, focal, task-specific dystonias are often associated with either dystonic or essential-type tremor. Such dystonic tremor occurs only during a specific action, and the tremor may not be evident when arms are outstretched in front of the body or when placed in any other position.
In children, the legs are often involved as the initial site of primary generalized dystonia; however, they are affected only rarely in adult dystonic patients. When dystonia affects the foot of an adult, one should consider the possibility of PD or a parkinsonian syndrome as the cause (11). The striatal foot deformity, with unilateral equinovarus dystonic posture of the foot and extension of the big toe (sometimes confused with Babinski’s sign), may be seen in as many as half of patients with PD. In addition to the striatal foot, some PD patients have a striatal hand deformity, often confused with rheumatoid arthritis (Fig. 28.10). We also have observed patients in whom foot or leg dystonia was the initial manifestation of the stiff-man syndrome, associated with positive antibodies against glutamic acid dehydrogenase (71).
< div class='tao-gold-member'>





