INTRODUCTION
The term “acanthocytosis” is derived from Greek to describe “thorny” red blood cells. The actual process resulting in the formation of acanthocytes is not known. Echinocytes, which are found under several pathologic conditions such as uremia, liver failure, and splenectomy, may show a spicular appearance with uniform discontinuous projections that are usually larger at their base than in their length, whereas the morphology of acanthocytes is characterized by a number of irregularly spaced thorny surface projections, often with terminal bulb (Fig. 44.1). Although echinocytes can normally comprise up to 3% of red blood cells in healthy adults, true acanthocytes should never be present in normal blood. For the purpose of avoiding false positives, due to experimental artifacts or echinocytes, it is suggested that the limit to be considered pathologic should be more than 3% crenated forms, and repeated sampling is required.
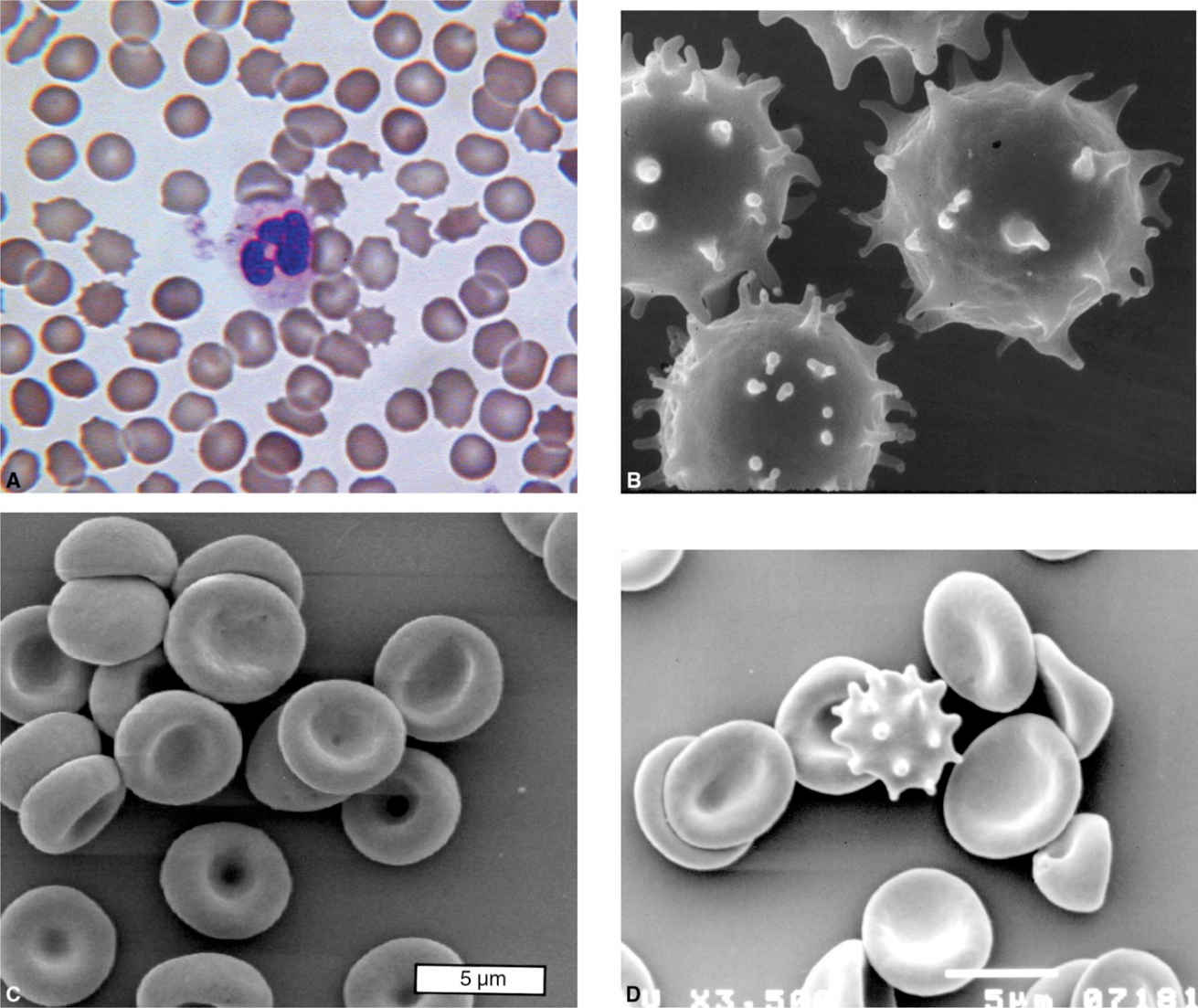
Figure 44.1. (A) Wet blood smear shows many acanthocytes with spiky projections in peripheral blood. (B) A scanning electron microscopic study shows irregularly shaped acanthocytes of ChAc (C) and NBIA (D) with several spicules of variable length compared to the control.

The terminology “neuroacanthocytosis” in the literature varies confusingly because not only it has been referred to by at least four different names (Levine–Critchley syndrome, acanthocytosis with neurologic disorder, choreoacanthocytosis, amyotrophic choreoathetosis with acanthocytosis) but also it contains at least five hereditary neurologic disorders (chorea–acanthocytosis [ChAc]: MIM200150, McLeod syndrome [MLS]: MIM300842, abetalipoproteinemia [ABL]: MIM200100, pantothenate kinase–associated neurodegeneration [PKAN]: MIM606157, Huntington disease–like 2 [HDL2]: MIM606438) (Table 44.1) (1). The responsible genes for these hereditary disorders with involvement of the nervous system and erythrocytes have been identified; however, molecular functions of the encoding proteins in human are still unclear. In this chapter, we focus our concentration on major neuroacanthocytosis (ChAc, MLS, ABL, and PKAN) based on reports of the cases with molecular diagnosis.
CHOREA–ACANTHOCYTOSIS
ChAc (MIM200150) is a hereditary devastating disease characterized by adult-onset, progressive hyperkinetic movements, and an unusual spiny erythrocyte morphology. This condition, first described in detail in the late 1960s, is a rare neurodegenerative disorder associated with acanthocytosis, so there have been no available data on the prevalence or incidence of ChAc (2,3). Although the disorder seems to be particularly common in Japan, there have been numerous reports from various countries (4). The responsible gene, VPS13A, was identified by two groups in 2001 (5,6).
CLINICAL FEATURES
The clinical features of ChAc are summarized in Table 44.2. The range of age at onset is between 6 and 62 years (mean around 35), though onset of symptoms is usually delayed until 25 to 45 years of age. Involuntary movement such as orolingual/facial dyskinesias, chorea, and tics are the most common, with chorea affecting the limbs, and sometimes the face, in almost all symptomatic patients (7,8). Choreic movements are enhanced by a command to stop the movements, mental calculation, or voluntary movements elsewhere in the body. Chorea is usually mild, and in some cases, dystonia is the dominant movement disorder. Severe involuntary movements affect the orofacial region, often sufficient to cause tongue and lip biting, and to interfere with speech and swallowing. Dystonia of the bulbar muscles often causes dysphagia and virtual anarthria, while abnormal palatal and pharyngeal contractions contribute to dysphagia. Coprolalia is rare. Dystonic protrusion of the tongue induced by mastication often forces food out of the mouth and exposes the tongue to being bitten. Persistent biting of the tongue, lips, or buccal mucosa is commonly found. Motor and vocal tics and dystonia were observed in one-half of the patients reported by Hardie et al. (7). Vocal tics consist of involuntary vocalization, including grunting, sucking, blowing, gasping, sighing, or monosyllabic utterances that are not recognizable words, and are often accompanied by belching, spitting, clicking, or sniffing. In addition to tics, ChAc patients often present with tourettism (3,9,10).
| Clinical Features of ChAc and MLS Patients with Genetic Diagnosis |
Findings | % in ChAca | % in MLSb |
Weak Kell antigen | 0 | 100 |
Acanthocytosis | 99 | 97 |
Involuntary movements | ||
Hyperkinesia face | 96 | 78 |
Chorea | 85 | 90 |
Self-mutilation | 94 | 38 |
Dystonia | 85 | 25 |
Parkinsonian features | 53 | 9 |
Cognitive and behavioral changes | ||
Cognitive changes | 70 | 59 |
Other psychiatric symptoms | 51 | 77 |
Dysarthria | 66 | 73 |
Dysphagia | 90 | 8 |
Elevation of serum creatine kinase | 96 | 94 |
Cerebellar dysfunction | 10 | 0 |
Neuromuscular manifestation | ||
Muscle weakness | 100 | 72 |
Muscle biopsy: myopathic/neuropathic | 25/50 | 77/64 |
Hyporeflexia/areflexia | 87 | 94 |
Sensory abnormality | 29 | 37 |
Seizure | 75 | 45 |
Cardiomyopathy | 2 | 68 |
aIncludes cases from References (3,11,16,18,27,65–78). | ||
Parkinsonism is relatively rare and only present at a progressive stage (7). Although parkinsonism rather than chorea can often be the predominant movement disorder, progressive parkinsonism usually gradually replaces the hyperkinetic state (7). Masked faces, dysarthria, dysphagia, and postural instability are characteristic, although generalized bradykinesia is uncommon, progressive supranuclear palsy, and apraxia of eyelid opening may present. In addition, axial symptoms including head drop or scoliosis are often observed (11). Cognitive impairment and behavioral changes are common, with cognitive impairment and psychosis manifesting as dementia, paranoia, and personality changes (7,8,12). Psychiatric symptoms may include depression, anxiety, and obsessive–compulsive behaviors. Impulsive and distracted behaviors or apathy with loss of insight is shown in some patients. In a neuropsychological review of 10 patients, 7 showed psychiatric abnormalities similar to those of Huntington’s disease (HD) (13). A frontal lobe–like syndrome may appear, and patients exhibit inefficiencies in psychometric tests of attention and planning. Some patients have impaired executive skills such as the ability to sustain concentration, to plan or change their behavior to reach a particular goal. Patients also tend to neglect personal appearance and social skills. They are observed in about half of the reported cases (7,8).
Epileptic seizures of temporal origin are considered as a predominant feature, because more than a half of ChAc patients are affected by seizures, which may precede other neurologic symptoms (14). The pathogenesis of the ChAc-associated seizure remains unclear; various studies with cranial magnetic resonance imaging (MRI) suggested that mesial temporal sclerosis would be a primary lesion of them (15).
Muscle weakness and atrophy are noted predominantly in the distal portion of the lower limbs in approximately a half of cases. Amyotrophy precedes clinical recognition of involuntary movements. Deep tendon reflexes shown are areflexia or hyporeflexia, and muscle tone is hypotonic in one-fourth of ChAc patients. There is no consistent sensory change. Splenomegaly, hepatomegaly, cardiomyopathy, pes cavus, cerebellar ataxia, and glove-stocking sensory loss are atypical.
GENETICS AND MOLECULAR MECHANISMS
In families, of diverse geographical origins, with ChAc having an autosomal recessive trait, the responsible gene for ChAc, VPS13A, has been identified on chromosome 9q21 and various mutations in autosomal recessive ChAc patients have been reported (5,6,16). The VPA13A gene is organized in 73 exons, and has two splicing variants, variant 1 (exons 1–68 and 70–73) and variant 2 (exons 1–69). There are more than 70 different mutations in VPS13A (8,16,17). Most mutations are nonsense or insertion/deletion mutations, some are changes affecting splicing, and a few mutations are missense mutations. This gene encodes a novel 3,096 amino acid protein chorein, which is localized to the Golgi apparatus and synaptosomes in the brain tissue. Also, it is expressed in the membrane fractures of erythrocytes, colocalizing in β-adducin and β-actin, suggesting in dysfunction there would be associated with the morphologic changes leading to acanthocytes (17–22). Also, various functional alterations of the ChAc erythrocytes (e.g., less endovesiculation and Ca2+ uptake) as well as aberrations in cytoskeletal organization have been detected (23,24).
NEUROPATHOLOGY AND NEUROCHEMISTRY
Postmortem studies of ChAc patients have revealed severe and extensive neuronal loss and gliosis in the caudate and putamen, globus pallidus. The thalamus is mildly affected, but the subthalamic nuclei, cerebral cortex, pons, medulla, and cerebellum are spared (7,25). The spinal cord is spared in most cases, and anterior horn cells and anterior and posterior roots are preserved, although some reports have described losses of upper or lower motor neurons (26). In the substantia nigra, neuronal loss is greatest in the ventrolateral region (27), whereas no neuronal loss of substantia nigra pars compact are detected in two siblings confirmed by loss of chorein expression by Western blotting (28). Histologically, both large and small neurons are depleted in the caudate. No amyloid or Lewy bodies have been reported.
Pathologic studies with nerve biopsy specimens have indicated the involvement affects primarily larger diameter myelinated fibers with segmental de- and remyelination in both motor and sensory nerves (7). Unmyelinated fibers are also depleted. Some reports have suggested that demyelination is secondary to axonal degeneration (29,30).
Muscle biopsies have disclosed only neuropathic changes, which contrasts with MLS in which both neuropathic and myopathic changes have been found to be present (8,31,32,). However, Ishikawa et al. (33) reported that CT scans of lower legs of ChAc patients revealed primary myopathic changes, which were not consistent with peripheral nerve distribution.
Neurochemical studies have shown the depletions of dopamine and its metabolites in most brain areas, particularly in the striatum. Noradrenaline levels in the globus pallidus and putamen are elevated, while substance P levels are decreased in the striatum and substantia nigra (34).
LABORATORY FINDINGS
Acanthocytes are found in peripheral blood smears of ChAc patients, though the percentage of acanthocytes is highly variable, usually being between 5% and 50%. A high percentage of acanthocytes does not correlate with disease severity. Chorein expression in ChAc patients’ erythrocyte is absent or reduced, which is the key investigation for the diagnosis of this disorder (20). Freeze–fracture electron microscopic studies have revealed ultrastructural abnormalities of the erythrocyte membranous skeleton.
Serum creatine kinase (CK) activity is commonly modestly elevated, although in some affected patients CK activity is within a normal range. Serum lipoprotein electrophoresis is normal in all subjects. Elevation in lactate dehydrogenase (LDH) and hepatic enzymes (aspartate aminotransferase, AST; alanine aminotransferase, ALT) are frequently observed but are not invariable. There is no reduction of serum cholesterol or of β-lipoprotein levels.
Nerve conduction velocity studies have shown a reduction of sensory nerve action potentials and prolongation of sensory conduction velocities in 25% to 50% of cases, but motor nerves are less affected. Electromyography studies have disclosed neurogenic changes with positive sharp waves and giant motor unit potentials, which are consistent with peripheral axonal pathology. Visual-evoked potentials, sensory-evoked potentials, and brain stem auditory-evoked potentials are normal.
Atrophy of the putamen and caudate nucleus with enlargement of lateral ventricles is typical findings of ChAc on CT and MRI (Fig. 44.2) (7,9,35). By use of MRI voxel-based morphometric technique, symmetric atrophy of the caudate head is specifically detected (36). These findings closely resemble those seen in HD. In contrast to HD, CT bicaudate index and frontal horn/bicaudate ratio in ChAc patients has not been shown to correlate with age, duration of illness, or severity of hyperkinesia. T2-weighed MRI also revealed high signal changes in the putamen and caudate, though these changes are not specific findings of ChAc (7,37,38). Positron emission tomography (PET) with various isotopes has shown bilateral symmetrically decreased glucose and oxygen metabolism in the caudate, putamen, and frontal cortex (Fig. 44.3) (10,38,39). PET with [18F]-fluorodopa has indicated decreased predopaminergic projection between the substantia nigra and the posterior putamen of affected patients, and PET with [11C]-raclopride revealed significant reduction of D2 postsynaptic receptors in the putamen and caudate (27,40). Single photon emission computed tomography (SPECT) has shown hypoperfusion in the frontal cortex and basal ganglia (see Fig. 44.3) (31,41).
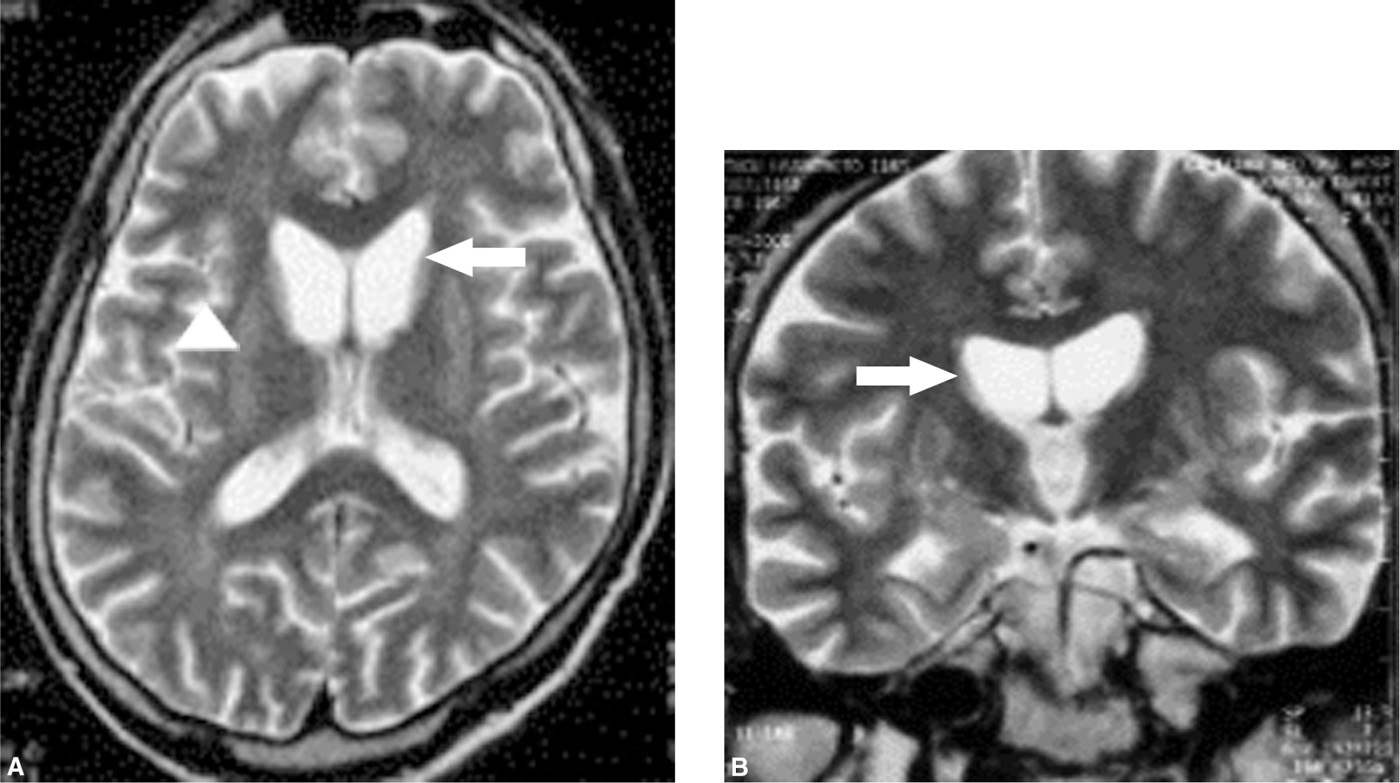
Figure 44.2. Axial (A) and coronal (B) T2-weighted images (1.5-T, Tr/TE=4500/9.6) show severe atrophy of the bilateral caudate nucleus (arrows), and heterogeneous intensity changes of the bilateral putamen (arrowhead).




