Encephalopathic Generalized Epilepsy and Lennox-Gastaut Syndrome
Kevin Farrell
William O. Tatum IV
Some generalized epilepsies are associated with cognitive dysfunction and encephalopathy. The clinical features of the seizures commonly observed in the encephalopathic generalized epilepsies were described in the 18th and 19th centuries (1). The development of electroencephalography demonstrated that patients with encephalopathic generalized epilepsy often had characteristic electroencephalographic (EEG) abnormalities. For instance, slow spike-and-wave discharges were observed in patients who often displayed brief motor seizures resistant to antiepileptic medications (2). As it became apparent that the natural history and response to treatment were often influenced by factors other than seizure type—for example, age of onset, intellectual function—the concept of epileptic syndromes became accepted and was embodied in the Classification of Epileptic Syndromes of the International League Against Epilepsy (3). In this classification, the term symptomatic generalized epilepsies was used for generalized epilepsies with encephalopathy and mental retardation. In 2001, a revision of the classification of both seizures and epileptic syndromes was proposed, suggesting that the term cryptogenic be replaced by probably symptomatic (4). Despite these proposals, many patients with encephalopathic generalized epilepsy do not fit clearly into a recognized syndrome. This chapter describes the clinical features, EEG abnormalities, response to treatment, and natural history of these epilepsies, including the Lennox-Gastaut syndrome, which is perhaps the best documented of these epilepsies.
SYNDROMES
Lennox-Gastaut Syndrome
The term Lennox-Gastaut syndrome (LGS) (5) has been used to describe a broad group of patients with (a) multiple generalized seizure types, typically tonic, atonic, myoclonic, and atypical absence; (b) an interictal EEG pattern characterized by diffuse slow spike-and-wave complexes; and (c) cognitive dysfunction, which develops in most but not all patients (5,6). Although cognitive dysfunction is common in patients with LGS (5,6), this often does not become apparent until later in the course of the disorder (5,7,8). Some authors have suggested that tonic seizures and paroxysmal fast rhythms at approximately 10 to 12 Hz during nonrapid eye movement (NREM) sleep are virtually never absent in LGS (7). Nonconvulsive status epilepticus is a common feature. The seizures are usually refractory to medical treatment.
The first seizure usually occurs between 1 and 8 years of age, with a peak between 3 and 5 years of age (8). The initial seizures may be partial or tonic-clonic, rather than the characteristic mixed motor seizures. Approximately 25% of children are neurologically normal prior to the onset of the epilepsy and have normal neuroimaging. In an additional 30% to 41% of patients, LGS is preceded by West syndrome (9,10). LGS occasionally occurs for the first time in the second decade of life (6,11). The onset of LGS in the second decade has also been described in patients who presented initially
with features of idiopathic generalized epilepsy, characterized by absence and generalized tonic-clonic seizures and by 3-Hz spike waves (6). In some of these patients, the clinical and EEG features of LGS developed after an episode of status epilepticus that was associated with an unsupervised withdrawal of antiepileptic medication, and treatment could be associated with seizure control and a disappearance of the slow spike-and-wave complexes (12). During adulthood, patients with partial epilepsy may develop drop attacks and slow spike-and-wave discharges that superficially resemble adult-onset LGS. Most of these patients probably have focal or multifocal epilepsy with secondary bilateral synchrony (8).
with features of idiopathic generalized epilepsy, characterized by absence and generalized tonic-clonic seizures and by 3-Hz spike waves (6). In some of these patients, the clinical and EEG features of LGS developed after an episode of status epilepticus that was associated with an unsupervised withdrawal of antiepileptic medication, and treatment could be associated with seizure control and a disappearance of the slow spike-and-wave complexes (12). During adulthood, patients with partial epilepsy may develop drop attacks and slow spike-and-wave discharges that superficially resemble adult-onset LGS. Most of these patients probably have focal or multifocal epilepsy with secondary bilateral synchrony (8).
It is clear that the term LGS is descriptive of a heterogeneous group of patients with differing etiologies and longterm outcomes. Categorizing a subgroup of patients with encephalopathic generalized epilepsy as LGS may add little toward treatment or prognosis and may have an inadvertent negative effect on parents and physicians, especially early in the course, by suggesting a very poor prognosis before longterm outcome is clear. Families who receive a diagnosis of LGS are often devastated when they research the issue independently, and in some cases, this dire interpretation is unwarranted. In other instances, a diagnosis of LGS may discourage physicians from continued aggressive efforts toward better seizure control. Although reference to LGS continues to appear in the remaining text, readers may want to reflect on the prudence of assigning the term LGS sparingly and instead use the broader term encephalopathic generalized epilepsy because of its greater phenotypic variability.
Other Encephalopathic Generalized Epilepsies
Many patients with multiple generalized seizure types and cognitive dysfunction do not demonstrate the generalized sharp and slow-wave complexes characteristic of LGS. A few of these patients have one of the syndromes described later in the differential diagnosis section. Most, however, are not amenable to syndromic diagnosis. Some are categorized by the dominant EEG abnormality. Thus, the multiple independent spike foci pattern on electroencephalograms is characterized by generalized motor seizures in most patients, with half of the patients having multiple seizure types, half having daily seizures, and two thirds being intellectually subnormal (13). With the advent of magnetic resonance imaging (MRI) and molecular genetics, many of these patients are now being categorized according to the underlying brain abnormality (e.g., hypothalamic hamartomas) (14) or molecular genetic disorder (e.g., mental retardation and seizures in males associated with mutation in the creatine-transporter gene located in Xq28) (15). The increasing ability to identify the underlying brain disease will clearly facilitate genetic counseling and also may prove very useful in counseling regarding the natural history of the epilepsy.
SEIZURE TYPES
Tonic, atypical absence, myoclonic, and atonic seizures are the characteristic seizure types in LGS and also are common in other encephalopathic generalized epilepsies. Tonic-clonic, clonic, or partial seizures may also be observed in patients with LGS and other encephalopathic generalized epilepsies, and are often the initial seizure type.
Tonic Seizures
Tonic seizures are the most common seizure type in LGS, particularly in patients with seizure onset at an early age (16). The prevalence of tonic seizures in LGS is between 74% and 90% in studies involving sleep EEG recordings (8). Mild episodes may be limited to a minimal upward deviation of the eyes (sursum vergens) and a brief respiratory disturbance. These mild seizures are common during sleep (17), and their clinical manifestations are often difficult to recognize without video-EEG recording (18,19). Tonic seizures may be restricted to neck flexors, and to facial and masticatory muscles; may involve the proximal muscles of the extremities, causing elevation and abduction of the arms; or may be associated with tonic involvement of the distal extremity muscles (8). Involvement of the extremities is often associated with a sudden fall, and these seizures may be difficult to distinguish from myoclonic or atonic seizures. Tonic seizures that last for longer than 20 seconds often have a vibratory component, with rapid and discrete clonic jerks. Such seizures are often mischaracterized as generalized tonic-clonic seizures. Altered consciousness may not be apparent for up to 1 second after onset of the EEG discharge, but recovery occurs within seconds of the end of the discharge (7). Gestural and ambulatory automatisms may follow the tonic phase, particularly in patients with late-onset LGS (20). Involvement of respiratory musculature is common and may result in apnea. Facial flushing, tachycardia, and pupillary dilation also have been described (21).
Atypical Absence Seizures
Atypical absence seizures are commonly seen in patients with LGS and other encephalopathic generalized epilepsies (7). They may be associated with eyelid myoclonias, mild tonic stiffening, autonomic features, and mild automatisms. Atypical absence seizures are not precipitated by photic stimulation. Gradual onset and offset, which are characteristic of atypical absence seizures, often make it difficult for caregivers to recognize these seizures (22).
Atonic Seizures
Atonic seizures may be difficult to distinguish clinically from myoclonias, myoclonic atonias, and tonic seizures, which may also cause the patient to fall (7). Studies
involving polygraphic recordings of muscle activity have demonstrated that drop seizures most often result from tonic seizures and that atonic seizures are less uncommon (23, 24, 25).
involving polygraphic recordings of muscle activity have demonstrated that drop seizures most often result from tonic seizures and that atonic seizures are less uncommon (23, 24, 25).
Myoclonic Seizures
Nonconvulsive Status Epilepticus
Nonconvulsive status epilepticus has been reported in 54% to 97% of patients with LGS, with higher rates observed in studies that included only patients with the classic features of LGS (29). Tonic seizures and confusion are the most common ictal manifestations (29). The confusional state may fluctuate and is often not recognized as a seizure, particularly in mentally retarded individuals. In patients with LGS, status epilepticus may last hours to weeks. In one study (30), the duration was longer than 1 week in 15 of 30 patients. Pure tonic status epilepticus occurs mostly in adults, is more frequent in patients whose initial seizures were tonic or tonic-clonic (30), and may be precipitated by intravenous (IV) administration of benzodiazepines (31, 32, 33, 34). Nonconvulsive status epilepticus may be inhibited by active wakefulness and facilitated by drowsiness; consequently, overmedication may compound the problem. The EEG pattern during status epilepticus may be difficult to distinguish from the interictal EEG pattern (29). The abnormalities include an increased amount of generalized slow spike-and-wave or polyspike-and-wave complexes, a hypsarrhythmia-like pattern, and 10-Hz discharges of variable duration (29).
ELECTROENCEPHALOGRAPHIC FEATURES
Interictal Electroencephalography
The interictal EEG background in LGS demonstrates a lower-than-normal frequency of the posterior dominant rhythm (35) and, generally, an increased amount of slow activity. Persistent background slowing is observed in 67% of patients and is associated with a poor prognosis for normal mental development (8). Background slowing is increased when the seizures are poorly controlled. Disturbances in NREM sleep patterns and total duration of REM sleep are observed in most patients with LGS who have severe mental retardation or frequent seizures (36), but these sleep features may be difficult to appreciate without a prolonged EEG recording and may be absent if the seizures are not frequent (37).
The interictal electroencephalogram in LGS is characterized by 2- to 2.5-Hz spike-and-wave and polyspike-and-wave discharges, although this pattern may be seen in other epilepsies as well. The spike-and-wave discharges are usually diffuse and maximal bifrontally, but are sometimes confined to the anterior or posterior head regions (38) (Fig. 27.1). The complexes are most often symmetrical, but focal or multifocal spikes and sharp waves are observed in 75% of patients, usually in the frontal or temporal regions (8). The slow spike waves are not induced by photic stimulation and are only rarely enhanced by hyperventilation (38). During slow-wave sleep, the discharges are more obviously bisynchronous and are often associated with polyspikes. Slow spike-wave complexes are often not present when the seizures initially occur, and in one series (16) they were first observed at a mean age of 44 months, in contrast to the mean age of seizure onset of 26 months. In later childhood, the slow spike-wave discharges gradually decrease in frequency or may disappear altogether.
Generalized paroxysmal fast activity manifesting as bursts of greater than 10-Hz rhythms, particularly during slow-wave sleep, are considered by some to be an integral feature of LGS (7,8) (Fig. 27.2). Nevertheless, this activity may not be present in all sleep recordings, and its prevalence depends on the duration of sleep recorded (39). The discharges are distributed diffusely, but are usually more prominent anteriorly and may be accompanied by tonic seizures. Polygraphic or video-EEG recordings may demonstrate subtle clinical signs that can be very brief and difficult to detect by observation alone.
The interictal EEG pattern in the other encephalopathic generalized epilepsies is variable. There is usually a poorly organized posterior dominant rhythm, and focal or multifocal abnormalities are common. Blume has described the clinical and EEG features in patients with multiple independent spike foci (13).
Ictal Electroencephalography
The ictal electroencephalogram during tonic seizures demonstrates bilaterally synchronous (10- to 25-Hz) activity maximal in the anterior and vertex regions, or attenuation of the background rhythm, which may be preceded by polyspikes, which may, in turn, be preceded or followed by generalized slow spike-and-wave activity. In contrast to generalized tonic-clonic seizures, postictal depression of EEG activity is uncommon. Tonic-automatic seizures are characterized by bilateral rapid discharges during the tonic phase and by diffuse slow spike-wave activity during automatism (7). The clinical features of a tonic seizure may not be apparent for up to 1 second after onset of the EEG discharge and may persist for several seconds after the discharge ends (40).
Atypical absence seizures are characterized by irregular, diffuse slow spike-and-wave discharges at approximately 2 to 2.5 Hz, which may be difficult to distinguish from the interictal slow spike-wave pattern. Rapid rhythms may be occasionally observed. Atonic and myoclonic seizures usually result in a loss of posture and may be difficult to detect without polygraphic video-EEG recordings. A variety of ictal abnormalities may be seen, including polyspike waves and, less commonly, slow spike waves or diffuse rapid rhythms (8).
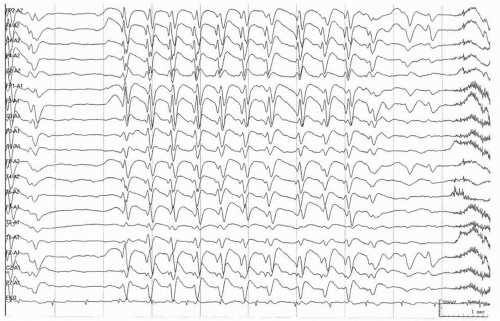 Figure 27.1 Interictal electroencephalogram demonstrates generalized sharp and slow-wave complexes. |
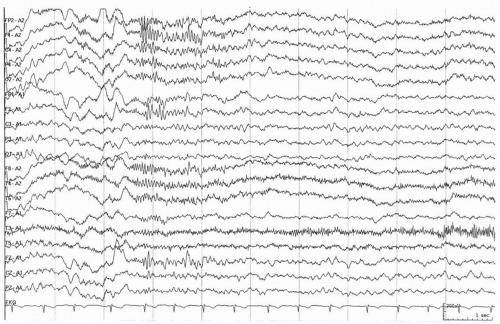 Figure 27.2 Paroxysmal fast activity occurs during nonrapid eye movement sleep without recognized clinical accompaniment. |
ASSOCIATED NEUROLOGIC DYSFUNCTION
In children with LGS, diffuse cognitive dysfunction is often not evident at seizure onset but becomes more apparent with time. In one series, 34% of patients had normal mental development at 4 years of age; however, 78% to 96% eventually developed mental retardation (41). Severe retardation occurs more often in patients with seizure onset before 3 years of age, frequent seizures, repeated episodes of status epilepticus, or symptomatic LGS, particularly in those with previous West syndrome (16,42). The cognitive dysfunction is exacerbated by several factors: the use of multiple medications, the tendency to increase the dose repeatedly because of poor seizure control, the effect of subtle seizures, and postictal confusion. Impaired information processing and prolonged reaction time are particularly prominent, with these patients consequently performing poorly on timed tests (43). Behavioral and personality disturbances, such as short attention span, aggressiveness, and disinhibition, complicate social adjustment and often confound attempts to integrate these individuals into the classroom and the community.
In children with LGS, motor development is affected less than cognitive development. Normal motor milestones were observed at 4 years of age in 79% of children with the disorder (41); however, many patients become ataxic or clumsy with time. Deterioration of gait over several years was a prominent feature in 17% of children in one series and, together with an increase in drop attacks, resulted in many becoming wheelchair-bound (44).
The degree of neurologic dysfunction observed in other encephalopathic epilepsies depends on the etiology and the severity of the underlying brain abnormality. Patients with secondary generalized epilepsy as a consequence of perinatal or postnatal brain injury are more likely to have spasticity.
ETIOLOGY
Congenital abnormalities of brain development are the most common cause of encephalopathic generalized epilepsy. They are demonstrated less often in LGS, and some abnormalities, such as Aicardi syndrome and lissencephaly, are rarely associated with LGS (5). In 20% to 30% of patients with LGS, there is normal psychomotor development at the onset, no history of brain injury, normal brain imaging, and indeterminate cause (8,16,45). Even in patients with LGS who have evidence of neurologic abnormality at presentation, the cause was determined in only 35 of 99 patients in one study (45). The most commonly reported abnormalities in LGS are congenital brain malformations, hypoxic-ischemic brain injury, encephalitis, meningitis, and tuberous sclerosis (5,45) (Table 27.1). Genetic and metabolic diseases are often associated with myoclonic seizures, but such conditions are rarely seen in LGS and are more often observed with other forms of encephalopathic generalized epilepsy. Mitochondrial disorders and neuronal ceroid lipofuscinosis are the most common genetic and metabolic disorders associated with encephalopathic generalized epilepsy. In individuals with encephalopathic epilepsy and absence of a developmental brain abnormality on head MRI scan, it is also important to evaluate the patient for possible aminoacidopathies, organic acidurias, and urea cycle disorders. Genetically determined diseases are increasingly being shown to be the pathologic basis of many cases of encephalopathic epilepsy. Genetic factors also play a role in LGS, with a family history of febrile seizures or epilepsy reported in 48% of patients with cryptogenic LGS (46). In addition, LGS has been reported to be a phenotype of generalized epilepsy with febrile seizures plus (GEFS+) (47).
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


