Figure 20.1. Diffuse slow spike-and-wave complexes on interictal EEG in a 7-year-old with LGS.
The finding of SSW on the EEG is an ominous finding. Drug-resistant generalized epilepsy is present in most patients with SSW (16). In one unselected group of patients with SSW on EEG, a >95% likelihood of manifesting seizures was predicted, with a >60% chance of having multiple seizure types, and a 70% chance of having difficult to control seizures (16).
It has been noted that children with interictal SSW discharges had underlying diffuse structural brain injury and subsequently a poor prognosis (16). Antecedent conditions associated with LGS almost always involve the cerebral cortex (1). Bilateral frontal lesions and diffuse dysplastic lesions of the cortex are commonly implicated. Interestingly, patients with Aicardi syndrome do not have LGS, suggesting that presence of the corpus callosum may be important (10). Lissencephaly is rarely associated with LGS (10). GPFA and tonic seizures are important because they help differentiate LGS from other epileptic encephalopathies that may have a better prognosis. The electrographic pattern of GPFA is a 10-Hz burst of bilateral fast activity that occurs during non–rapid eye movement sleep (Fig. 20.2). These bursts are generally brief and may appear frequently during sleep and disappear during rapid eye movement sleep (10). They are identical to the discharges seen with tonic seizures but may have minimal clinical signs such as brief apnea or mild axial tonic features that are best illustrated on electromyography. GPFA is not pathognomonic for LGS since it may also occur in localization-related epilepsy with focal seizures that arise from extratemporal (often frontal lobe) origins. Patients with EGE, tonic seizures as the predominant seizure type, and GPFA on EEG had a worse prognosis than did patients presenting with predominantly atypical absence, myoclonic or atonic seizures (17).
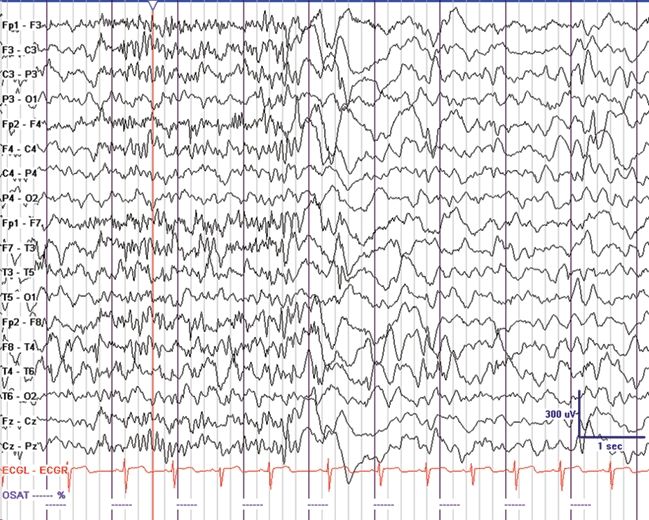
Figure 20.2. GPFA in a 4-year-old with EGE and mixed seizures. Tonic seizures were noted in sleep with eye opening, mild axial stiffening, and apnea. (Courtesy of Joseph Casadonte, MD.)
DIFFERENTIAL DIAGNOSIS
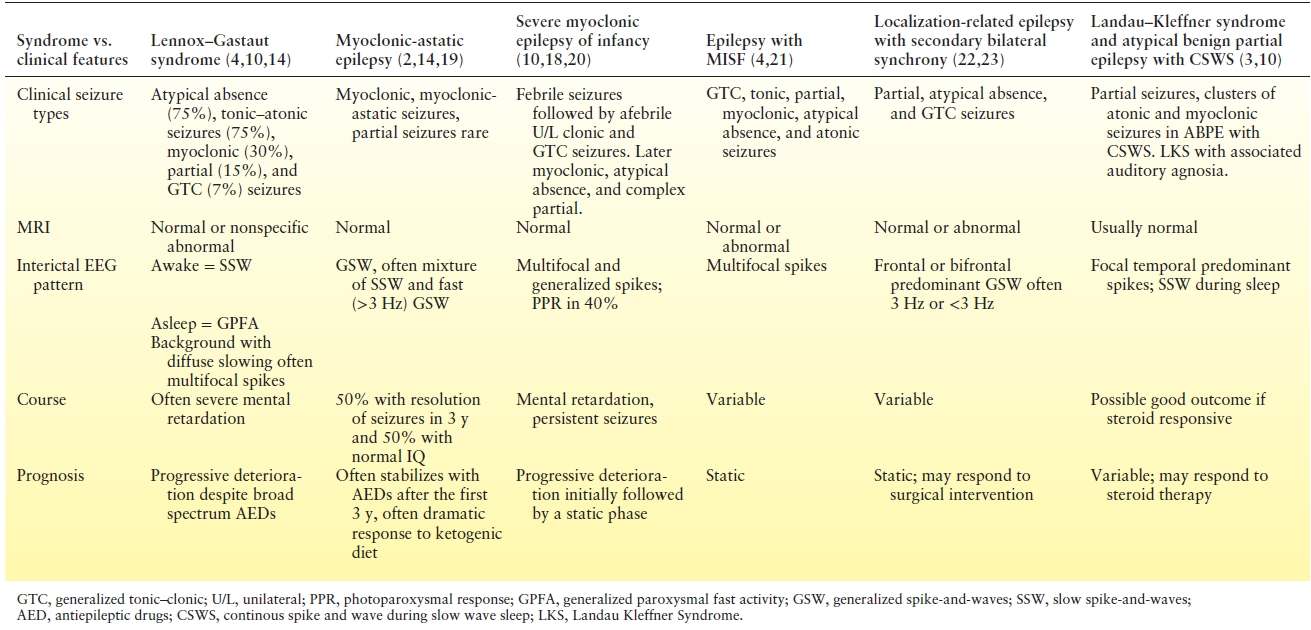
Most of the epileptic encephalopathies evolve over the first few years after the clinical presentation. In contrast to LGS, certain epileptic encephalopathies, such as severe myoclonic epilepsy of infancy (SMEI) (aka Dravet syndrome) and epilepsy with MISF, rarely have SSW (4,18). If SSW are intermixed with 3-Hz or faster GSW, then genetic generalized epilepsy should be considered, especially in patients with normal mental status. If there are predominantly SSW during sleep, and focal interictal electrical discharges while awake, then atypical benign partial epilepsy with continuous SSW of sleep is an important differential diagnosis. Focal abnormalities on neuroimaging, EEG, clinical semiology, and physical examination may suggest localization-related epilepsy with focal seizures and SBS on EEG when intermixed features are encountered that are not state specific. This is particularly important as resective surgery in localization-related epilepsy may be a treatment option when patient have focal seizures in the setting of bilateral EEG abnormalities or SBS. The most difficult diagnostic distinction is between MAE and LGS (Table 20.1).
Table 20.1 Summary of the Encephalopathic Generalized Epilepsy Syndromes and Clinical Features
West Syndrome
WS is present in 2 to 5 per 100,000 children and consists of a triad that includes ES, developmental arrest, and a unique EEG abnormality referred to as hypsarrhythmia (see Chapter 16) (Fig. 20.3). Hypsarrhythmia is an interictal chaotic, very high-voltage, asynchronous background with intermixed multifocal spikes and sharp waves that is associated with ES. Hypsarrhythmia in the setting of WS may precede LGS and may later on evolve into a SSW pattern (see Chapter 17) (see Fig. 20.3). A history of ES may occur in up to 40% of children with LGS though variable EEG patterns may be evident (1,6,8,9). Parallel associations among ES, MISF, and LGS exist. For example, patients with Down syndrome may have ES and hypsarrhythmia on the EEG that transition to EGE with MISF or to LGS in approximately 5% of patients (24). The onset of WS is usually between 4 and 7 months of age including 90% that are associated with neurologic abnormalities from underlying structural, genetic, or inborn errors of metabolism present (25).
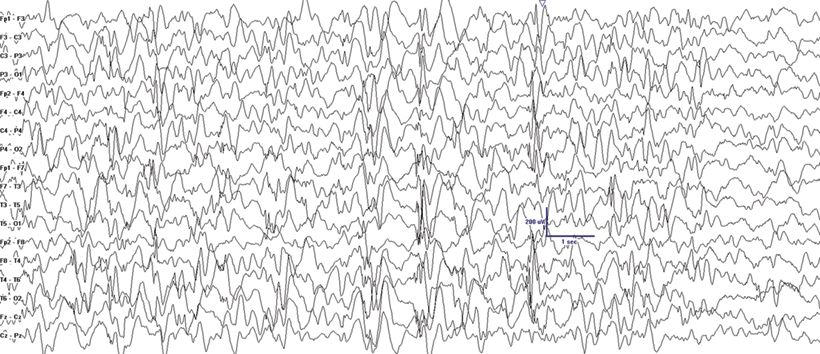
Figure 20.3. Hypsarrhythmia depicted on ictal EEG in a 7-month-old with new-onset epileptic spasms. A 2-week course of ACTH was followed by resolution of hypsarrhythmia and spasms.
Myoclonic Astatic Epilepsy
MAE was first described by Doose as a childhood EGE syndrome characterized by seizure types that are similar to those seen with LGS including atypical absence, myoclonic, atonic, tonic, and tonic–clonic seizures (2). In contrast to LGS, MAE has more prominent myoclonic seizures and less prominent tonic seizures early in the course of the condition. Focal seizures are rare. MAE usually presents between 7 months and 6 years of age. The characteristic massive myoclonic seizures are brief symmetrical jerks involving the neck, shoulders, and arms followed by an abrupt loss of muscle tone that usually results in a fall (astatic seizure). Patients with MAE may also present with pure atonic seizures, atypical absence, GTC seizures, or with episodes of nonconvulsive SE. Most patients are neurologically normal at onset, and there is a family history of epilepsy in 40% of patients (2). The ictal EEG shows spike- or polyspike-and-wave discharges at a frequency of 2 to 4 Hz. The interictal EEG may be normal. When it is abnormal, it may show brief bursts of 3-Hz GSW, 4- to 7-Hz parietal theta, and 4-Hz occipital slowing consistently blocked by eye opening (Fig. 20.4) (26). In contrast to the uniformly poor prognosis of LGS, seizures are controlled in at least half of the patients with MAE within 3 years, and more than 50% of patients will have a normal IQ in this subgroup (14). Valproic acid (VPA), lamotrigine (LTG), and ethosuximide have been generally used for treatment (14). Carbamazepine, phenytoin, and vigabatrin may worsen minor motor seizures. Many centers now consider the ketogenic diet (KD) after failure of one to two drugs or even as a first-line therapy because of its often superior seizure control in this syndrome (20).
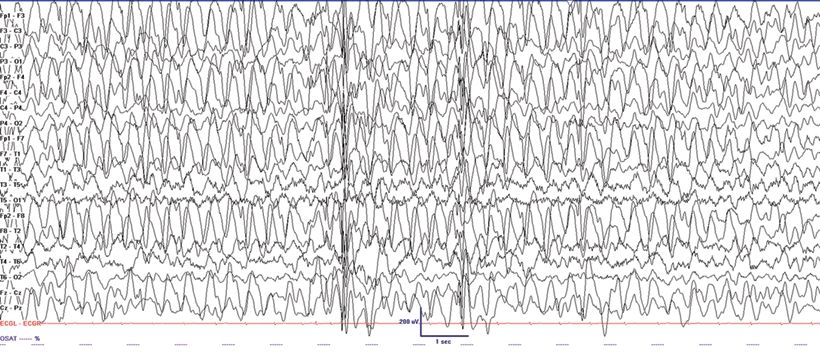
Figure 20.4. Generalized spike-and-wave at varying frequencies in a 2-year-old patient with MAE. Treatment with KD normalized the EEG.
Dravet Syndrome
SMEI also known as Dravet syndrome is a childhood epileptic encephalopathy that usually presents with prolonged febrile seizures during the first year of life. Febrile hemiclonic or GTC seizures are common presentations (18). During the second and third years of life, myoclonic seizures and atypical absences subsequently appear. The myoclonic seizures may be massive and associated with falls. Tonic–clonic and focal seizures are also common. Tonic seizures are rare, and when present, they are infrequent. The EEG is often initially normal but may show SSW as an ictal correlate during atypical absence seizures. Over time, focal abnormalities may develop in addition to generalized spike- and polyspike-and-wave discharges that are usually associated with myoclonic seizures. Stimulus-provoked seizures can occur, particularly light or stress induced, and therefore testing for photosensitivity may be useful (18). Although the myoclonic seizures may resolve, other seizure types persist, and there is progressive cognitive deterioration and overall poor developmental prognosis, with ongoing difficult-to-control seizures. After age 5 years, GTC seizures with nocturnal predominance are usually the predominant seizure type. Behavioral disorders are common. Ataxia is frequently seen, and adolescents may develop a peculiar crouched gait (18). As genetic techniques have improved, patients have shown SCN1A mutations in approximately 70% to 80% of Dravet patients, with 95% of these mutations occurring de novo (27). There is a wide spectrum of epilepsies associated with mutations of SCN1A including generalized epilepsy with febrile seizures plus (GEF+). SCN1A mutations are estimated to cause 7% of severe epilepsies in children under the age of 3 years. The mutation is suspected to cause dysfunction of interneurons involved in the inhibitory circuit of the brain (18). Mortality in SMEI is estimated to be 15% by age 20. SUDEP is well described in this disease (18). Treatment is difficult, and VPA, levetiracetam (LEV), zonisamide (ZNS), topiramate (TPM), and clobazam (CLB) are often used. Stiripentol, in conjunction with VPA and/or CLB, has been shown to be helpful in reducing frequency of seizures (28). A Dravet-like phenotype with a better prognosis is also seen in patients with mutations of protocadherin 19 (PCDH19) in females (29).
Early-Onset Epileptic Encephalopathies
Early infantile epileptic encephalopathy (EIEE) or Ohtahara syndrome accounts for only 0.2% of the early childhood epilepsies. It presents with drug-resistant tonic epileptic spasms, burst suppression on EEG, and has an overall poor prognosis. Seizure onset is within the first 3 months of life and asymmetric tonic seizures and focal seizures occur in approximately one-third of patients. EIEE often evolves to WS between 3 and 6 months of age and subsequently into LGS between 1 and 3 years of age (30). The cause is often symptomatic, and a high mortality rate is encountered in infancy. It generally is associated with malformations of cortical development. Another early epileptic encephalopathy is EME. It presents in the first month with vigorous myoclonias and multifocal seizures. Both EIEE and EME patients later in the course of development manifest tonic seizures and with a burst suppression pattern on EEG. Differences between the two entities include tonic seizures as the main type of seizures in EIEE, in addition to an earlier onset, and the presence of burst suppression in both the awake and sleep state (30). The similarity of the EEG patterns has led authors to question whether EIEE and EME are separate entities (22). DNA sequencing has led to identification of genes that can help define a genetic basis for some of the early-onset epileptic encephalopathies (EIEE, EME, and WS).
Atypical Benign Partial Epilepsy with Continuous Spike-and-Wave of Sleep
This syndrome presents with developmental stagnation or regression and includes features of EGE and localization- related epilepsy. Due to developmental decline and often medical intractability during the acute stage when presenting with continuous spike-and-wave in slow sleep, it has also been termed “pseudo-LGS” (10). Atypical benign partial epilepsy with continuous spike-and-waves in sleep may present with clusters of myoclonic and atonic seizures but not tonic seizures between 2 and 6 years of age. The seizures cluster lasting 2 to 4 weeks and then are separated by seizure-free periods (3). This differentiates this syndrome from LGS where tonic seizures are common and remissions do not occur. The EEG shows diffuse SSW discharges during sleep (Fig. 20.5). Central spikes are usually prominent in this syndrome similar to other forms of more benign focal epilepsy. Remission occurs before age 15, but patients may be left with a permanent cognitive impairment (3).
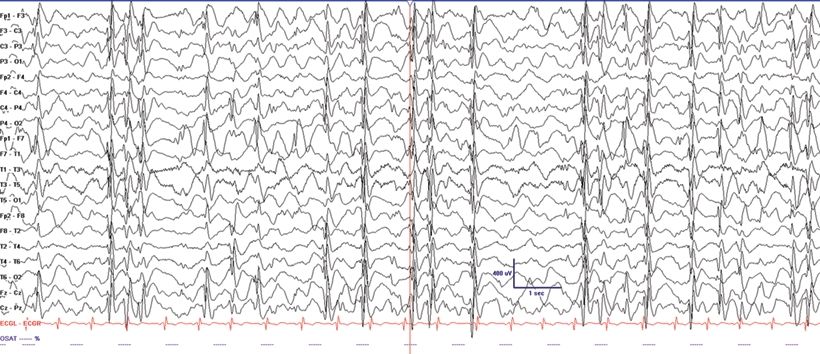
Figure 20.5. Electrical SE in slow-wave sleep in a 7-year-old child with Landau–Kleffner syndrome.
Localization-Related Epilepsy with Secondary Bilateral Synchrony
Focal epilepsy with SBS is usually present when there is a focal EEG spike that leads into a generalized discharge resembling SSW. Because of the limitations of surface scalp EEG recordings, there may be variability in the presence of a preceding focal discharge before the generalized burst in seen. Characterization of the discharges shows that they tend to have a frontal predominance at 2 to 2.5 Hz. The interval between the focal discharge and the onset of the generalized discharge is very brief but is greater than the mean callosal transmission time and may further decrease in duration as the discharge continues (5). Since the primary focus may not be discernable on routine EEG, it may mimic a generalized discharge. Foci in the frontal lobes are most often associated with this phenomenon, but this fast spread of an initially focal discharge can also be seen elsewhere along the midline of the brain (21). Furthermore, clinical seizures associated with this phenomenon may imitate absence seizures further providing a challenge to differentiate a generalized from focal seizure.
Severe Epilepsy with Multiple Independent Spike Foci
EGE with MISF has been proposed as an independent entity (31) that is clinically related to LGS due to associated MR and drug-resistant seizures. Three or more epileptic foci are present with at least one in each hemisphere. In patients with this EEG finding, more than 50% of patients had more than one type of seizure and 50% were having daily seizures (4). This is particularly true for patients presenting with spikes at least every 10 seconds (4). When EGE with MISF is present, a variety of seizures including GTC, focal, tonic, myoclonic, atypical absence, and atonic seizures may be seen. It has extensive overlap with the other epileptic encephalopathies and may frequently transition in an age-dependent fashion to other epilepsies including LGS, EIEE, and WS (31). It may also be seen in up to 20% of symptomatic LGS patients as they age, but rarely in those without a structural–metabolic basis (32). Like LGS, EGE with MISF may be seen in patients with extensive bilateral cerebral pathology and manifests as severe drug-resistant epilepsy. When the EEG features of MISF are present, a more variable prognosis with a larger potential for normal cognitive development is present, and up to one-third of patients may be developmentally normal (4).
Genetic Syndromes Causing EGE
Angelman syndrome (AS) is characterized by severe developmental delay, absent speech, paroxysms of laughter, a puppet- like gait with ataxia, and jerky movements (also referred to as “happy puppet syndrome” in the past although some consider this term now obsolete), in addition to other distinctive clinical features. Patients with AS also have intractable epilepsy and EEG characteristics that may be confused with LGS (33). Seizure types that may be observed include atypical absence, myoclonic, clonic, and complex partial seizures. Characteristic EEG findings are diffuse, bilateral, frontally predominant, high-amplitude delta waves with a notched or triphasic-like slow waves that mimic SSW complexes. The notched appearance of the sharp waves superimpose upon the slower delta activity. The frequency may be 2 to 2.5 Hz and is maximal in the prefrontal derivations, similar to the SSW seen in LGS (Fig. 20.6). Due to the notched appearance of sharp waves, the findings have also been referred to as “ill-defined slow spike-and-wave” (33). The 10-Hz GPFA seen in LGS is not seen in AS (33).
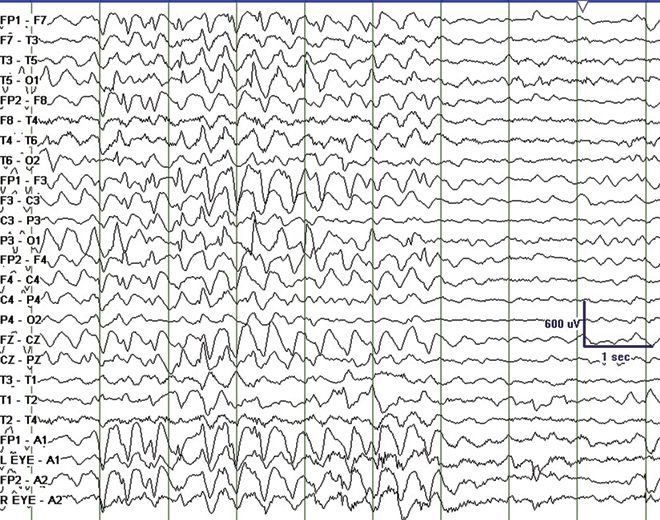
Figure 20.6. “Ill-defined” slow spike-and-wave on interictal EEG in a 4-year-old patient with Angelman syndrome.(Courtesy of Maria Gieron-Korthals, MD.)
Rett syndrome is encountered between 6 months and 3 years of age and occurs only in females. Epilepsy in typical Rett syndrome usually presents after age 3 years (34). Rett syndrome is characterized by initial normal development followed by cognitive regression, autistic features, microcephaly, ataxia, and hand-wringing movements in addition to multiple seizure types that include absence, myoclonic, and atonic seizures mimicking LGS. The EEG may show progressively worsening slowing of the background activity with needle-like central spikes that are activated by somatosensory stimulation. A unique feature includes a 4- to 6-Hz rhythmic theta pattern maximal centrally (35). Rett Syndrome may also present with SSW (Fig. 20.7). Paroxysmal nonepileptic events are common in patients with Rett syndrome, and video EEG can be helpful to distinguish epileptic and nonepileptic events. There are atypical variants in which epilepsy often presents in infancy without initial normal development. In typical Rett syndrome, there is an associated mutation in the MECP2 gene at Xp28.2 in 93% of cases. In atypical variants, MECP2 mutations are responsible for 50% to 70%. Other causes include MECP2 duplications, CDKL5 mutations, and FOXG1 mutations (34).
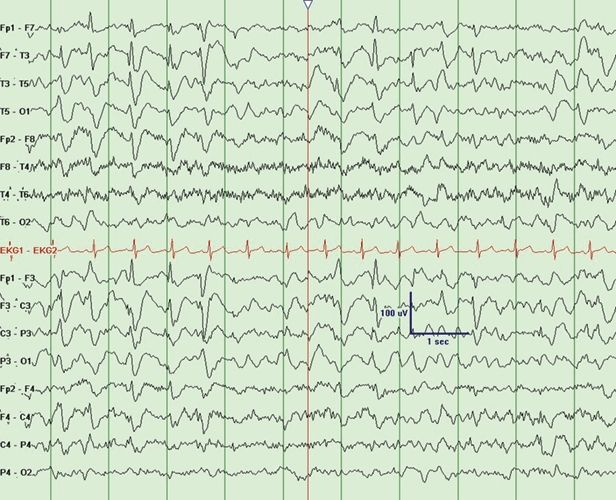
Figure 20.7. Slow spike-and-wave in an 8-year-old child with Rett syndrome.(Courtesy of Selim R. Benbadis, MD.)
Related posts:
 Epidemiologic Aspects of Epilepsy
Application of Electroencephalography in the Intensive Care Setting
Hormones, Catamenial Epilepsy, Sexual Function, and Reproductive and Bone Health in Epilepsy
Other Nonepileptic Paroxysmal Disorders
Driving and Social Issues in Epilepsy
Dietary Therapies for Epilepsy
Epidemiologic Aspects of Epilepsy
Application of Electroencephalography in the Intensive Care Setting
Hormones, Catamenial Epilepsy, Sexual Function, and Reproductive and Bone Health in Epilepsy
Other Nonepileptic Paroxysmal Disorders
Driving and Social Issues in Epilepsy
Dietary Therapies for Epilepsy
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


