Chapter 26 Endocrine Physiology in Relation to Sleep and Sleep Disturbances
Modulation of Endocrine Function by Sleep–Wake Homeostasis and Circadian Rhythmicity
In healthy adults, reproducible changes of essentially all hormonal and metabolic variables occur during sleep and around wake–sleep and sleep–wake transitions. These daily events reflect the interaction of central circadian rhythmicity and sleep–wake homeostasis. Thus, the dual control of sleep timing and quality by circadian processes (i.e., Process C) and sleep–wake homeostasis (i.e., Process S) is readily reflected in the modulatory effects exerted by sleep on endocrine and metabolic function. Pathways by which central circadian rhythmicity and sleep-wake homeostasis affect peripheral endocrine function and metabolism include the modulation of the activity of the hypothalamic releasing and inhibiting factors and the autonomous nervous system control of endocrine organs. Findings from genome-wide association studies also support a role of circulating melatonin levels on specific endocrine targets, including the pancreatic beta cells.1–3 The relative contributions of circadian timing compared with homeostatic control in the regulation of the temporal organization of hormone release vary from one endocrine axis to another. Similarly, modulatory effects of the transitions between wake and sleep (and vice versa) and between non–rapid eye movement (NREM) and rapid eye movement (REM) stages also vary from one hormone to another.
Circadian oscillations can be generated in many peripheral organs, including tissues that release endocrine signals such as adipocytes and pancreatic beta cells.4 These “local” oscillators appear to be under the control of the central pacemaker in the suprachiasmatic nuclei either directly via neural and endocrine signals, or indirectly via its control of behavioral rhythms such as the sleep–wake cycle and the rhythm of feeding. The possible involvement of these peripheral oscillators on the temporal organization of endocrine release and metabolic function during waking and sleeping remains to be investigated.
To differentiate between effects of circadian rhythmicity and those subserving sleep–wake homeostasis, researchers have used experimental strategies that take advantage of the fact that the central circadian pacemaker takes several days to adjust to a large sudden shift of sleep–wake and light–dark cycles (such as occur in jet lag and shift work). Such strategies allow for the effects of circadian modulation to be observed in the absence of sleep and for the effects of sleep to be observed at an abnormal circadian time. Figure 26-1 illustrates mean profiles of hormonal concentrations, glucose levels, and insulin-secretion rates (ISR) observed in healthy subjects who were studied before and during an abrupt 12-hour delay of the sleep–wake and dark–light cycles. To eliminate the effects of feeding, fasting, and postural changes, the subjects remained recumbent throughout the study, and the normal meal schedule was replaced by intravenous glucose infusion at a constant rate.5
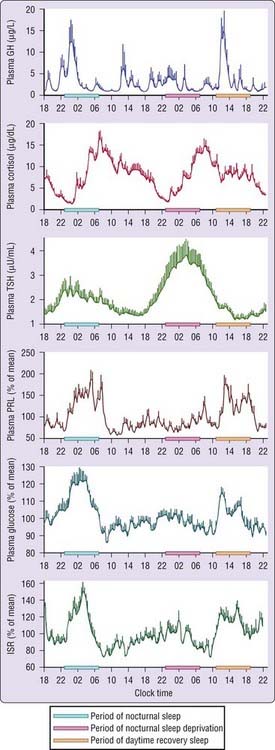
As shown in Figure 26-1, this drastic manipulation of sleep had only modest effects on the wave shape of the cortisol profile, in sharp contrast with the immediate shift of the growth hormone (GH) and prolactin (PRL) rhythms that followed the shift of the sleep–wake cycle. The temporal organization of thyroid-stimulating hormone (TSH) secretion appears to be influenced equally by circadian and sleep-dependent processes. Indeed, the evening elevation of TSH levels occurs well before sleep onset and has been shown to reflect circadian phase. During sleep, an inhibitory process prevents TSH concentrations from rising further. Consequently, in the absence of sleep, the nocturnal TSH elevation is markedly amplified. Both sleep and time of day clearly modulated glucose levels and ISR. Nocturnal elevations of glucose and ISR occurred even when the subjects were sleep deprived, and recovery sleep at an abnormal circadian time was also associated with elevated glucose and ISR. This pattern of changes in glucose levels and ISR reflected changes in glucose use because, when glucose is infused exogenously, endogenous glucose production is largely inhibited.
First we will review the interactions between sleep and endocrine release in the hypothalamic-pituitary axes and the roles of sleep in carbohydrate metabolism, appetite regulation, and hormone control of body-fluid balance. Table 26-1 provides basic information about the hormones that will be discussed in this chapter. We then summarize the growing body of evidence linking decrements of sleep duration or quality that occur with sleep restriction, in sleep disorders, or as a result of normal aging, and disturbances of endocrine and metabolic function. Lastly, we review recent evidence linking disorders of sleep–wake regulation and metabolic and endocrine diseases, including obesity, type 2 diabetes, and polycystic ovary syndrome (PCOS). For a review of sleep abnormalities in other endocrine diseases, the reader is referred to Chapter 125.
Table 26-1 Origin and Main Action of Hormones
| HORMONE | MAIN SECRETING ORGAN | MAIN ACTION IN ADULTS |
|---|---|---|
| Growth hormone (GH) | Pituitary gland | Anabolic hormone that regulates body composition |
| Prolactin (PRL) | Pituitary gland | Stimulates lactation in women; pleiotropic actions |
| Adrenocorticotropic hormone (ACTH) | Pituitary gland | Stimulates release of cortisol from adrenal cortex |
| Cortisol | Adrenal cortex | Stress hormone, antiinsulin effects |
| Thyroid-stimulating hormone (TSH) | Pituitary gland | Stimulates the release of thyroid hormones from the thyroid gland |
| Luteinizing hormone (LH) | Pituitary gland | Stimulates ovarian and testicular function |
| Follicle-stimulating hormone (FSH) | Pituitary gland | Stimulates ovarian and testicular function |
| Testosterone | Gonads | Stimulates spermatogenesis |
| Estradiol | Ovaries | Stimulates follicular growth |
| Insulin | Pancreas | Regulates blood glucose levels |
| Melatonin | Pineal gland | Hormone of the dark that transmits information about the light–dark cycle |
| Leptin | Adipose tissue | Satiety hormone regulating energy balance |
| Ghrelin | Stomach | Hunger hormone regulating energy balance |
The Growth Hormone Axis
Pituitary release of GH is stimulated by hypothalamic GH-releasing hormone (GHRH) and inhibited by somatostatin. In addition, the acylated form of ghrelin, a peptide produced predominantly by the stomach, binds to the growth hormone secretagogue (GHS) receptor and is a potent endogenous stimulus of GH secretion.6 There is a combined and probably synergic role of GHRH stimulation, elevated nocturnal ghrelin levels, and decreased somatostatinergic tone in the control of GH secretion during sleep. Although sleep clearly involves major stimulatory effects on GH secretion, the hormones of the somatotropic axis, including GHRH, ghrelin, and GH, in turn appear to be involved in sleep regulation.7
In healthy adult subjects, the 24-hour profile of plasma GH levels consists of stable low levels abruptly interrupted by bursts of secretion. The most reproducible GH pulse occurs shortly after sleep onset.8 In men, the sleep-onset GH pulse is generally the largest, and often the only, secretory pulse observed over the 24-hour span. In women, daytime GH pulses are more frequent, and the sleep-associated pulse, although still present in the vast majority of individual profiles, does not account for the majority of the 24-hour secretory output. Sleep onset elicits a pulse in GH secretion whether sleep is advanced, delayed, or interrupted and reinitiated. The mean GH profile shown in Figure 26-1 illustrates the maintenance of the relationship between sleep onset and GH release in subjects who underwent a 12-hour delay shift of the sleep–wake cycle. There is a consistent relationship between the appearance of delta waves in the EEG and elevated GH concentrations and maximal GH release occurs within minutes of the onset of slow-wave sleep (SWS).8,9 In healthy young men, there is a quantitative correlation between the amount of GH secreted during the sleep-onset pulse and the duration of the slow-wave episode.10 Pharmacologic stimulation of SWS with oral administration of low doses of gamma-hydroxybutyrate (GHB), a drug used for the treatment of narcolepsy, as well as with ritanserin, a selective 5-hydroxytryptamine (5-HT2) receptor antagonist, results in increases in GH secretion.11,12 Sedative hypnotics that are ligands of the GABAA receptor such as benzodiazepines and imidazopyridines, do not increase nocturnal GH release, consistent with their lack of stimulation of slow-wave activity.13
The mechanisms that underlie the relationship between early sleep and GH release are unclear. The significance of this relationship is that anabolic processes in the body are synchronized to a state when behavioral rest occurs and when cerebral glucose use is at its lowest point.14 There is good evidence to indicate that stimulation of nocturnal GH release and stimulation of SWS reflect, to a large extent, synchronous activity of at least two populations of hypothalamic GHRH neurons.14 Sleep-onset GH secretion appears to be primarily regulated by GHRH stimulation occurring during a period of decreased somatostatin inhibition of somatotropic activity. Indeed, in humans, GH secretion during early sleep may be nearly totally suppressed by administration of a GHRH antagonist.15 The late evening and nocturnal hours coincide with the trough of a diurnal variation in hypothalamic somatostatin tone16 that is likely to facilitate nocturnal GH release. It is also possible that ghrelin plays a role in causing increased GH secretion during sleep because the normal 24-hour ghrelin profile shows a marked nocturnal increase peaking in the early part of the night.17,18
The upper panel of Figure 26-1 shows that the secretion of GH is increased during sleep independently of the circadian time when sleep occurs and that sleep deprivation results in greatly diminished release of this hormone. However, a slight increase may be observed during nocturnal sleep deprivation, indicating the existence of a weak circadian component that could reflect, as discussed earlier, lower somatostatin inhibition. Following a night of total sleep deprivation, GH release is increased during the daytime such that the total 24-hour secretion is not significantly affected.19 Again, the mechanisms underlying this compensatory daytime secretion are unknown, but they could involve decreased somatostatinergic tone or elevated ghrelin levels.
Marked rises in GH secretion before the onset of sleep have been reported by several investigators.20–22 Presleep GH pulses may reflect the presence of a sleep debt, as they occur consistently after recurrent experimental sleep restriction.23 The short-term negative feedback inhibition exerted by GH on its own secretion may also explain observations of an absent GH pulse during the first slow wave period, when a secretory pulse occurred before sleep onset. Awakenings interrupting sleep have an inhibitory effect on GH release.24,25 Thus, sleep fragmentation generally decreases nocturnal GH secretion.
The Corticotropic Axis
The mean cortisol profile shown in Figure 26-1 illustrates the remarkable persistence of this diurnal variation when sleep is manipulated. Indeed, the overall waveshape of the profile is not markedly affected by the absence of sleep or by sleep at an unusual time of day. Thus, the 24-hour periodicity of corticotropic activity is primarily controlled by circadian rhythmicity.
Nevertheless, modulatory effects of sleep or wake have been clearly demonstrated. Indeed, a number of studies have indicated that sleep onset is reliably associated with a short-term inhibition of cortisol secretion,5,26 although this effect may not be detectable when sleep is initiated at the time of the daily maximum of corticotropic activity, that is, in the morning.27 Under normal conditions, because cortisol secretion is already quiescent in the late evening, this inhibitory effect of sleep, which is temporally associated with the occurrence of slow-wave sleep,28–30 results in a prolongation of the quiescent period. Therefore, under conditions of sleep deprivation, the nadir of cortisol secretion is less pronounced and occurs earlier than under normal conditions of nocturnal sleep. Conversely, awakening at the end of the sleep period is consistently followed by a pulse of cortisol secretion.5,25,31
During sleep deprivation, the rapid effects of sleep onset and sleep offset on corticotropic activity are obviously absent, and, as may be seen in the profiles shown in Figure 26-1, the nadir of cortisol levels is higher than during nocturnal sleep (because of the absence of the inhibitory effects of the first hours of sleep), and the morning maximum is lower (because of the absence of the stimulating effects of morning awakening). Overall, the amplitude of the rhythm is reduced by approximately 15% during sleep deprivation as compared to normal conditions.
In addition to the immediate modulatory effects of sleep–wake transitions on cortisol levels, nocturnal sleep deprivation, even partial deprivation, results in an elevation of cortisol levels on the following evening.32 Sleep loss thus appears to delay the normal return to evening quiescence of the corticotropic axis. This endocrine alteration is remarkably similar to that occurring in normal aging, where increases in evening cortisol levels of similar magnitude are consistently observed. This interpretation is consistent with findings in normal subjects submitted to recurrent partial sleep restriction, as discussed later.
The Thyroid Axis
Daytime levels of plasma TSH are low and relatively stable and are followed by a rapid elevation starting in the early evening and culminating in a nocturnal maximum occurring around the beginning of the sleep period.30,33 The later part of sleep is associated with a progressive decline in TSH levels, and daytime values resume shortly after morning awakening. The first 24 hours of the study illustrated in Figure 26-1 are typical of the diurnal TSH rhythm. Because the nocturnal rise of TSH occurs well before the time of sleep onset, it probably reflects a circadian effect. However, a marked effect of sleep on TSH secretion may be seen during sleep deprivation (clearly seen in Fig. 26-1), when nocturnal TSH secretion is increased by as much as 200% over the levels observed during nocturnal sleep. Thus, sleep exerts an inhibitory influence on TSH secretions, and sleep deprivation relieves this inhibition.30,34
Interestingly, when sleep occurs during daytime hours, TSH secretion is not suppressed significantly below normal daytime levels. Thus, the inhibitory effect of sleep on TSH secretion appears to be operative when the nighttime elevation has taken place, indicating once again the interaction of the effects of circadian time and the effects of sleep. When the depth of sleep at the habitual time is increased by prior sleep deprivation, the nocturnal TSH rise is more markedly inhibited, suggesting that SWS is probably the primary determinant of the sleep-associated fall.30 Awakenings interrupting nocturnal sleep appear to relieve the inhibition of TSH and are consistently associated with a short-term TSH elevation.
Circadian and sleep-related variations in thyroid hormones have been difficult to demonstrate, probably because these hormones are bound to serum proteins and thus their peripheral concentrations are affected by diurnal variations in hemodilution caused by postural changes. However, under conditions of sleep deprivation, the increased amplitude of the TSH rhythm may result in a detectable increase in plasma triiodothyronine (T3) levels, paralleling the nocturnal TSH rise.35 If sleep deprivation is continued for a second night, then the nocturnal rise of TSH is markedly diminished as compared with that occurring during the first night.35,36 It is likely that following the first night of sleep deprivation, the elevated thyroid hormone levels, which persist during the daytime period because of the prolonged half-life of these hormones, limit the subsequent TSH rise at the beginning of the next nighttime period. Data from a study of 64 hours of sleep deprivation suggest that prolonged sleep loss may be associated with an upregulation of the thyroid axis, with lower levels of TSH and higher levels of thyroid hormones.37 As discussed in the section on chronic sleep restriction, this was indeed the case in a study of the endocrine and metabolic effects of a sleep debt resulting from bedtime curtailment to 4 hours per night for 6 nights.38,39
The inhibitory effects of sleep on TSH secretion are time dependent, and that may cause, under specific circumstances, elevations of plasma TSH levels that reflect the misalignment of sleep and central circadian timing. In a study examining the course of adaptation to an abrupt 8-hour advance of the sleep–dark period in healthy young men,40 TSH levels increased progressively because daytime sleep failed to inhibit TSH and nighttime wakefulness was associated with large circadian-dependent TSH elevations. As a result, mean TSH levels following awakening from the second shifted sleep period were more than twofold higher than during the same time interval following normal nocturnal sleep. This overall elevation of TSH levels was paralleled by a small increase in T3 concentrations.40 This study demonstrated that the subjective discomfort and fatigue often referred to as “jet-lag syndrome” are associated not only with a desynchronization of bodily rhythms but also with a prolonged elevation of a hormone concentration in the peripheral circulation.
Prolactin Secretion
Under normal conditions, PRL levels undergo a major nocturnal elevation starting shortly after sleep onset and culminating around midsleep. Decreased dopaminergic inhibition of PRL during sleep is likely to be the primary mechanism underlying this nocturnal PRL elevation. In adults of both sexes, the nocturnal maximum corresponds to an average increase of more than 200% above the minimum level.35 Morning awakenings and awakenings interrupting sleep are consistently associated with a rapid inhibition of PRL secretion.35
Studies of the PRL profile during daytime naps or after shifts of the sleep period have consistently demonstrated that sleep onset, irrespective of the time of day, has a stimulatory effect on PRL release. This is well illustrated by the profiles shown in Figure 26-1, in which elevated PRL levels occur both during nocturnal sleep and during daytime recovery sleep, whereas the nocturnal period of sleep deprivation was not associated with an increase in PRL concentrations. However, the sleep-related rise of PRL may still be present, although with a reduced amplitude, when sleep does not occur at the normal nocturnal time. Maximal stimulation is observed only when sleep and circadian effects are superimposed.41–43 A close temporal association between increased prolactin secretion and slow wave activity is apparent.44 However, in contrast to the quantitative correlation between amount of slow wave activity and amount of GH release that has been evidenced in men, no such “dose-response” relationship has been demonstrated for prolactin. Awakenings interrupting sleep inhibit nocturnal PRL release.44
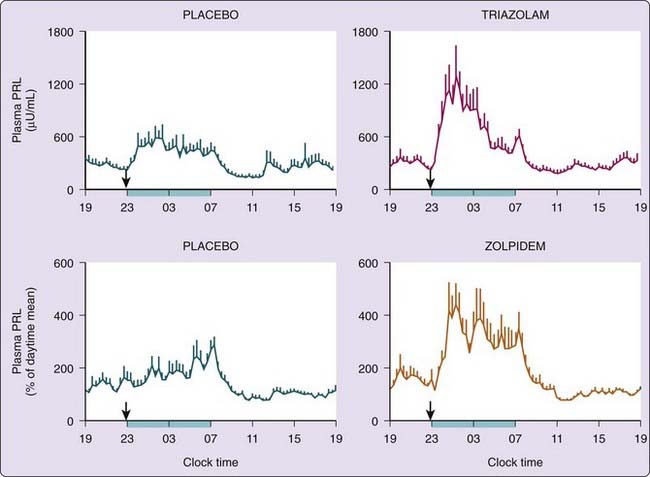
Benzodiazepine and imidazopyridine hypnotics taken at bedtime may cause an increase in the nocturnal PRL rise, resulting in concentrations near the pathological range for part of the night.45,46 This is illustrated for triazolam and zolpidem in Figure 26-2. Neither triazolam nor zolpidem has any effect on the 24-hour profiles of cortisol, melatonin, or GH. A recent report showed that chronic treatment of insomnia with the melatonin receptor agonist ramelteon also increases prolactin release in women, but not in men.47
There is evidence from animal studies that PRL is involved in the humoral regulation of REM sleep.48 The primary effect is a stimulation of REM sleep, which appears to be dependent on time of day. Recent findings indicate that prolactin deficient mice have decreased REM sleep.49
The Gonadal Axis
The relationship between the 24-hour patterns of gonadotropin release and gonadal steroid levels varies according to the stage of maturation, and it is gender dependent in young adulthood (for review see Van Cauter et al.35).
In adult men, the day–night variation of plasma LH levels is dampened or even undetectable. During the sleep period, LH pulses appear to be temporally related to the REM–NREM cycle.50 Despite the low amplitude of the nocturnal increase in gonadotropin release, a marked diurnal rhythm in circulating testosterone levels is present, with minimal levels in the late evening, a robust rise following sleep onset and maximal levels in the early morning.51,52 Thus, the robust circadian rhythm of plasma testosterone may be partially controlled by factors other than LH. The nocturnal rise of testosterone appears temporally linked to the latency of the first REM episode,53 as plasma levels continue to rise until the first REM episode occurs. A robust rise of testosterone may also be observed during daytime sleep, suggesting that sleep, irrespective of time of day, stimulates gonadal hormone release.54 Experimental sleep fragmentation in young men resulted in attenuation of the nocturnal rise of testosterone, particularly in subjects who did not achieve REM sleep.55 Androgen concentrations in young adults decline significantly during periods of total sleep deprivation and recover promptly once the sleep is restored.54,56 In contrast, pharmacological suppression of testosterone in healthy men appears to have no effect on the total amount and overall architecture of nighttime sleep.57
In women, the 24-hour variation in plasma LH is markedly modulated by the menstrual cycle.58,59 In the early follicular phase, LH pulses are large and infrequent, and a marked slowing of the frequency of secretory pulses occurs during sleep, suggestive of inhibitory effect of sleep on pulsatile gonadotropin-releasing hormone (GnRH) release. Awakenings interrupting sleep are usually associated with a pulse of LH concentration.60 In the midfollicular phase, pulse amplitude is decreased, pulse frequency is increased, and the frequency modulation of LH pulsatility by sleep is less apparent. Pulse amplitude increases again by the late-follicular phase. In the early luteal phase, the pulse amplitude is markedly increased, the pulse frequency is decreased, and nocturnal slowing of pulsatility is again evident. In the mid- and late-luteal phase, pulse amplitude and frequency are decreased, and there is no modulation by sleep.
In older men, the amplitude of LH pulses is decreased but the frequency is increased and no significant diurnal pattern can be detected.61–63 The circadian variation of testosterone persists, although markedly dampened.63 The sleep-related rise is still apparent in older men, but its amplitude is lower and the relationship to REM latency is no longer apparent.64 It is likely that decreased sleep quality as occurs in aging as well as in sleep disorders (e.g., obstructive sleep apnea) plays a role in the dampening of the sleep-related testosterone rise.
In postmenopausal women, gonadotropin levels are elevated, but they show no consistent circadian pattern.65 A number of studies66–68 have indicated that estrogen replacement therapy has modest beneficial effects on subjective and objective sleep quality, particularly in the presence of environmental disturbance69 or sleep-disordered breathing.66,67,70
Glucose Regulation
The consolidation of human sleep in a single 7- to 9-hour period implies that an extended period of fast must be maintained overnight. Despite the prolonged fasting condition, glucose levels remain stable or fall only minimally across the night. In contrast, if subjects are awake and fasting in a recumbent position, in the absence of any physical activity, glucose levels fall by an average of 0.5 to 1.0 mmol/L (± 10 to 20 mg/dL) over a 12-hour period.71 Thus, a number of mechanisms that operate during nocturnal sleep must intervene to maintain stable glucose levels during the overnight fast.
The lower panels of Figure 26-1 show profiles of blood glucose and ISR obtained in normal subjects who were studied under conditions of constant glucose infusion,76 a condition that results in a marked inhibition of endogenous glucose production. Thus, when subjects receive a constant glucose infusion, changes in plasma glucose levels reflect mainly changes in glucose use. A marked decrease in glucose tolerance (reflected in higher plasma glucose levels) is apparent during nighttime as well as daytime sleep. A smaller elevation of glucose and insulin also occurs during nocturnal sleep deprivation, indicating an effect of circadian-dependent mechanisms.
Under conditions of constant glucose infusion, the decrease in glucose tolerance during the first half of the sleep period is reflected in a robust increase in plasma glucose, which is followed by a more than 50% increase in insulin secretion. It is estimated that about two thirds of the fall in glucose use during sleep is due to a decrease in brain glucose metabolism72 related to the predominance of slow wave stages, which are associated with a 30% to 40% reduction in cerebral glucose metabolism as compared to the waking state.73 (See Chapter 18.) The last third of the fall would then reflect decreased peripheral use. Diminished muscle tone during sleep and rapid antiinsulin-like effects of the sleep-onset GH pulse are both likely to contribute to decreased peripheral glucose uptake. The nocturnal elevation of melatonin levels could contribute to the nocturnal decrease in glucose tolerance because of an inhibitory effect of melatonin on insulin release from beta cells.2,74 During the later part of the sleep period, glucose levels and insulin secretion decrease to return to presleep values, and this decrease appears to be partially due to the increase in wake and REM stages.75 Indeed, glucose use during the REM and wake stages is higher than during NREM stages.73 In addition, several other factors may also contribute to the decline of glucose levels during late sleep. These include the hypoglycemic activity of previously secreted insulin during early sleep, the increased insulin-independent glucose disposal due to transient mild hyperglycemia, and the quiescence of GH secretion and thus the rapid attenuation of the short-term inhibitory effects of this hormone on tissue glucose uptake. Finally, the later part of the night appears to be associated with increased insulin sensitivity, reflecting a delayed effect of low cortisol levels during the evening and early part of the night.76
Sleep and Appetite Regulation
Sleep plays an important role in energy balance. In rodents, food shortage or starvation results in decreased sleep77 and, conversely, total sleep deprivation leads to marked hyperphagia.78 The identification of hypothalamic excitatory neuropeptides, referred to as hypocretins or orexins, that have potent wake-promoting effects and stimulate food intake, has provided a molecular basis for the interactions between the regulation of feeding and sleeping.79,80 Orexin-containing neurons in the lateral hypothalamus project directly to the locus coeruleus and other brainstem and hypothalamic arousal areas, where they interact with the leptin-responsive neuronal network involved in balancing food intake and energy expenditure. Orexin-containing neurons are active during waking and quiescent during sleep. Orexin activity is inhibited by leptin, a satiety hormone, and stimulated by ghrelin, an appetite promoting hormone.
Leptin, a hormone released by the adipocytes, provides information about energy status to regulatory centers in the hypothalamus.81 Circulating leptin concentrations in humans show a rapid decline or increase in response to acute caloric shortage or surplus, respectively. These changes in leptin levels have been associated with reciprocal changes in hunger. The 24-hour leptin profile shows a marked nocturnal rise, which is partly dependent on meal intake.82 The upper panel of Figure 26-3 shows a typical 24-hour profile of plasma leptin levels in a normal man. The nocturnal elevation of leptin has been thought to suppress the hunger during the overnight fast. Although daytime food intake plays a major role in the nocturnal rise of leptin, a study using continuous enteral nutrition to eliminate the impact of meal intake showed the persistence of a sleep-related leptin elevation, though the amplitude was lower than during normal feeding conditions.83 Prolonged total sleep deprivation results in a decrease in the amplitude of the leptin diurnal variation.84
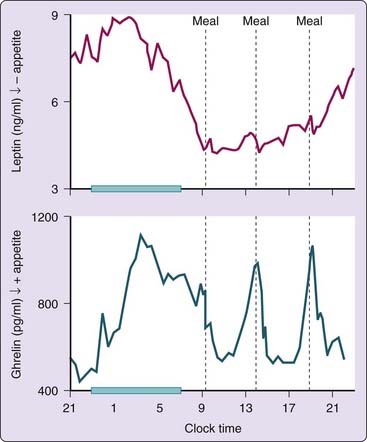
Ghrelin is also involved in regulating energy balance6 and stimulating appetite.85 Daytime profiles of plasma ghrelin levels are primarily regulated by the schedule of food intake: Levels rise sharply before each designated meal time and fall to trough levels within 1 to 2 hours after eating. A study examining spontaneous meal initiation in the absence of time- and food-related cues provided good evidence for a role for ghrelin in meal initiation.86 The 24-hour profile of ghrelin levels shows a marked nocturnal rise, which is only modestly dampened when subjects are sleep deprived.17 The nocturnal ghrelin rise partly represents the rebound of ghrelin following the dinner meal. Despite the persistence of the fasting condition, ghrelin levels do not continue to increase across the entire sleep period and instead decrease during the later part of the night. The lower panel of Figure 26-3 illustrates a representative 24-hour profile of ghrelin from a normal subject who ingested three carbohydrate-rich meals.
Water and Electrolyte Balance During Sleep
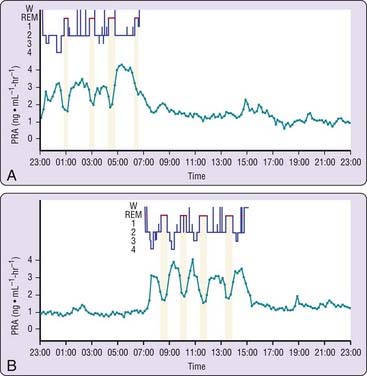
Vasopressin release is pulsatile but without apparent relationship to sleep stages.87 Levels of atrial natriuretic peptide are relatively stable and do not show fluctuations related to the sleep–wake or REM–NREM cycles.88 Whether the levels of plasma atrial natriuretic peptide exhibit a circadian variation is still a matter of controversy.88 A close relationship between the beginning of REM episodes and decreased activity has been consistently observed for plasma renin activity.87,89–91 Figure 26-4 illustrates the 24-hour rhythm of plasma renin activity in a subject studied during a normal sleep–wake cycle and in a subject studied following a shift of the sleep period. A remarkable synchronization between decreased plasma renin activity and REM stages is apparent during both sleep periods.92 This relationship was confirmed in studies with selective REM-sleep deprivation in healthy subjects.93

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


