Figure 23.1. Functional anatomy of the hypothalamic-pituitary-adrenal axis.
* The target gland hormones also work as feedback hormones (negative feedback) at the hypothalamic and pituitary levels.
Hypothalamic regulatory hormone | Target gland/ secreted feedback hormone | Major physiologic effects of pituitary hormones | Clinical findings of pituitary hormone deficiency | |
Anterior pituitary hormone | ||||
ACTH | CRH, ADH | Adrenal/cortisol | Several vital functions via cortisol such as vascular integrity and blood pressure maintenance, response to stress, immunomodulatory effects | Weight loss, weakness, dizziness, vomiting, circulatory collapse, fever, shock, hypoglycemia, hypotension, hyponatremia |
GH | GHRH, somatostatin | Multiorgan/IGF-I from liver | Direct effects or via IGF-I. Promote growth, control protein, lipid and carbohydrate metabolism, control memory function | Adults: visceral obesity, fatigue, impairment of attention and memory, premature atherosclerosis |
TSH | TRH | Thyroid/T4,T3 | Control energy metabolism and vital functions | Tiredness, cold intolerance, dry skin, cognitive slowing, bradycardia, hypotension |
Gonadotropins (FSH/LH) | GnRH | Ovary and testis/E2, T | Control reproductive function | Infertility, decreased libido, deceased muscle mass, osteoporosis |
Prolactin | PIF (Dopamine) | Breast | Milk production, immunomodulatory effects? | Agalactia |
Posterior pituitary hormone | ||||
ADH | – | Kidney | Conservation of body water and plasma osmolality | Polyuria, polydipsia, hypernatremia |
Oxytocin | – | Breast and uterine | Stimulates milk ejection and uterine contraction | ? |
Table 23.2. Components of the hypothalamic-pituitary-adrenal axis: hormones secreted by the pituitary gland, hypothalamus and target organs. Major physiologic effects of pituitary hormones and the clinical findings of pituitary hormone deficiencies.
ACTH = adrenocorticotropin hormone; ADH = antidiuretic hormone; CRH = corticotropin-releasing hormone; E2 = estradiol; FSH = follicle-stimulating hormone; GH = growth hormone; GHRH = growth hormone-releasing hormone; GnRH = gonadotropin-releasing hormone; IGF = insulin-like growth factorLH = luteinizing hormone; PIF = prolactin release-inhibitory factor; T = testosterone; T3=triiodothyronine; T4 = thyroxine; TRH = thyrotropin-releasing hormone; TSH = thyroid-stimulating hormone.
All anterior pituitary hormones are secreted in a pulsatile fashion and tend to follow a diurnal pattern. Antidiuretic hormone (ADH; vasopressin) is produced by the supraoptic and paraventricular nuclei of hypothalamus and travels in the axons through the pituitary stalk to the posterior pituitary gland. The chief physiologic stimulus of ADH secretion is an increase in serum osmolality and a decrease in plasma volume, resulting in water reabsorption at the level of the distal and collecting ducts of the kidney. Small increments in serum osmolality >290 mOsm/kg induce prompt secretion of ADH.
The blood supply of the hypothalamus is provided by the small branches of the arteries of the circle of Willis. Arterial supply to the anterior pituitary gland, median eminence, and stalk is derived from the superior hypophyseal arteries. These arteries form the primary vascular plexus and converge into venules to form the long and short hypophyseal portal veins. These veins descend to the pars tuberalis and pars distalis of the anterior lobe, where a secondary plexus of sinusoidal capillaries is formed. The long portal veins pass through the diaphragma sella, thus being vulnerable to mechanical compression from brain or pituitary swelling or direct stalk injury. In contrast, the short portal veins originate below the diaphragma sella.
The anterior pituitary lobe, particularly its lateral aspects, receives its blood supply indirectly after passage through the median eminence and portal vessels. Any interruption of the portal vessels may result in anterior pituitary dysfunction. In contrast, the neurohypophysis receives its direct arterial blood supply from the inferior hypophyseal arteries.
23.1.3 Endocrinology of Normal Stress Response
The physiologic response to stress involves the activation of the stress regulatory centres in the central nervous system, which, in turn, activates the HPA axis and the autonomic nervous system. The brain, hippocampus and amygdala in particular, are closely involved in the stress response. The stress response is predominantly regulated by the sympathoadrenal system, which includes the sympathetic nervous system and the adrenal medulla, and by the HPA axis. The stress system also influences other hypothalamo-pituitary axes (those controlling gonadal, thyroidal, and growth functions) and exerts complex effects on immune/inflammatory reactions. The end hormones of the neuroendocrine system, particularly glucocorticoids and catecholamines, act to maintain behavioural, cardiovascular, metabolic, and immune homeostasis during stress.
Severe acute stress, as occurs in critically ill patients, leads to strong activation of the HPA axis. Activation of the HPA axis involves increased secretion of corticotropin-releasing hormone (CRH) and antidiuretic hormone (ADH), also called arginine vasopressin, which enhances CRH activity. CRH stimulates the production of adrenocorticotropin hormone (ACTH), causing the adrenal cortex to produce more cortisol and dehydroepiandrosterone. Additionally, CRH activates the sympathoadrenal system at the same time. Following road accidents or during major surgery or sepsis, the human body can increase its glucocorticoid production by 5- to 10-fold. An adequate increase in glucocorticoid production is therefore crucial for survival and is required to cope with the severe stress during critical illness.
23.1.4 General Principles of Pituitary Hormone Changes During the Acute Phase of Brain Injury
Alterations in pituitary function during the acute phase of brain injury result in a temporary increase or decrease in pituitary hormone concentrations in blood circulation. Recent data revealed differences between alterations in hypothalamic-pituitary function in the acute phase (the first hours to days) and those in the prolonged phase or chronic phase (from 10 to 15 days beyond) of critical illness. During the acute phase, the secretory pattern of pituitary hormones inactivates anabolic target hormones. This response has been suggested to be beneficial and adaptive. In prolonged critical illness when the patient continues under intensive medical care, the pulsatile secretion of the anterior pituitary hormones generally decreases because of decreased hypothalamic stimulation, and this underlies the impaired anabolism in critical care patients.
The normal pituitary gland responds to acute brain injury with two sorts of secretory patterns: increased production of ACTH, prolactin (PRL) and growth hormone (GH), and decreased or unchanged production of gonadotropins (FSH and LH), and thyroid-stimulating hormone (TSH), associated with a decreased activity of their target organ. Changes in circulating hormone levels become apparent during the first hours or days after trauma and may persist for the duration of the acute critical illness. These hormonal alterations reflect the acute adaptive response to the injury and may be influenced by the type of injury and pharmacological therapy given to treat the critical illness (glucocorticoids, narcotic analgesics or dopaminergic agents). The basic philosophy of the neuroendocrine response after acute brain injury may be summarized as follows: these adaptations are initially protective for the human body; however, if they are inadequate, as in pituitary dysfunction or excessive, they may be harmful and cause endocrine, metabolic and immune disturbances. Therefore, the most important target for intensive care should be to differentiate adaptive response from pathological hormonal changes and to avoid unnecessary interventions.
23.1.5 Anterior Pituitary Dysfunction in the Acute Phase of Brain Injury
The Hypothalamo-pituitary-adrenal Axis
The HPA axis status is the most extensively investigated endocrine function in critically ill brain-injured patients (Table 23.1). Patients normally present with increased cortisol levels. These changes may persist up to 10 to 15 days after brain injury and are essential for maintaining vascular tone and endothelial integrity. Initially, cortisol increase is mediated by ACTH. Later, other substances, mainly cytokines and catecholamines, act as mediators. The diurnal variation of cortisol may be preserved or abolished.
TBI studies investigating the association between cortisol dynamics, severity of TBI (assessed by the Glasgow Coma Scale, GCS) and outcome prediction have yielded controversial results. Some investigators concluded that higher cortisol values are associated with more severe head trauma and with a worse clinical outcome, whereas others found lower cortisol values in the most severely injured patients with brainstem dysfunction or in brain-dead victims. Still other studies found no correlations between baseline cortisol and TBI severity. Acute adrenal failure after TBI may occur as a consequence of ischemia or direct trauma to components of the HPA axis, a secondary form of adrenal insufficiency. In addition, primary failure of the adrenal gland due to a systemic inflammatory response after the release of pro-inflammatory mediators may complicate recovery from TBI. The only difference between primary adrenal failure and secondary adrenal failure is the presence of mineralocorticoid deficiency. However, because the common clinical problem is due to hypocortisolism, it is not practical to differentiate primary from secondary adrenal failure in routine clinical management, commonly called adrenal failure. Throughout the text, HPA axis insufficiency and adrenal failure are used interchangeably.
According to clinical studies, the incidence of adrenal failure after acute TBI is 10-50% (Table 23.1). The incidence of HPA axis insufficiency during the acute phase is generally 20-25%, which signals a high risk. Although the data are heterogeneous, critically ill patients with nontraumatic brain injury may also experience adrenal failure. Recent studies suggest that around 25-30% of patients with ischemic stroke or aneurysmal subarachnoid hemorrhage have HPA axis insufficiency during the acute phase (Table 23.1).
Current evidence suggests that HPA axis insufficiency immediately after brain injury is associated with poor neurological outcome, greater need for vasoactive drug therapy, hyponatremia, relative or absolute hypocalcemia, hemodynamic instability, and rapidly progressive hypotension, all of which may increase the risk of death. Therefore, such clinical signs/symptoms should prompt their urgent investigation as a possible cause of adrenal insufficiency during acute care.
Algorithm for Diagnostic Testing and Management
The adequacy of cortisol secretion in critical illness, including brain injury, remains a diagnostic challenge because the normal range for baseline or stimulated cortisol levels in critically ill patients is inadequately defined. Recent published guidelines recommend acute anterior pituitary hormone evaluation in all patients with moderate-to-severe TBI, but those with documented baseline skull fracture, increased intracranial pressure or diabetes insipidus require specific attention. There are no defined selection criteria for patients with nontraumatic brain injury; however, in all brain-injured patients the best selection criteria for endocrine evaluation, particularly of the HPA axis, are clinical findings (Table 23.2) and high clinician suspicion.
Serum basal cortisol measurement is the primary test to evaluate the HPA axis. Although a morning cortisol test is recommended by some authors, stress will decrease the normal diurnal variation in cortisol release, making random values acceptable. Cortisol will be low in most cases of both primary and secondary adrenal failure. The actual concentration that defines a low value is somewhat controversial because of the expected increase in cortisol in response to brain-injury-induced stress. The amount of cortisol required cannot be predicted from the severity of injury and it may change as the clinical status changes. Although some authors suggest random basal cortisol levels <25 µg/dl or 36 µg/dl, a cortisol concentration <15 µg/dl (410 nmol/l) are acceptable as a diagnostic value for adrenal insufficiency in critically ill brain-injured patients. In patients lower values and clinical findings consistent with adrenal insufficiency, there is no need for further stimulation testing in routine clinical practice.
ACTH may also be directly measured to differentiate primary from secondary adrenal failure. As mentioned, differentiation between primary and secondary adrenal failure is not warranted in critically ill brain-injured patients in routine clinical practice. Additionally, because ACTH measurement is not routinely available in most hospital laboratories, urgent measurement is not recommended.
In patients with basal cortisol levels between 15 and 25 µg/dl and suspicious clinical findings, then ACTH stimulation testing is warranted. Standard-dose ACTH stimulation test (after blood sampling for baseline cortisol measurement, 250 µg ACTH is administered intravenously and cortisol measured at 30 and/or 60 minutes after stimulation) is routinely used in the diagnosis of primary or secondary adrenal insufficiency. In normal conditions, peak cortisol response after ACTH stimulation >20 µg/dl is defined as adequate cortisol response. In critically ill brain-injured patients, however, the best cut-off value after standard ACTH stimulation test is yet to be defined. The recommended value of 25 µg/dl (690 nmol/l) seems more realistic as a cut-off higher than that used in an outpatient setting because the peaks achieved during this test are, on average, higher than in patients without stress or severe illness.
In some studies, low-dose (1 µg) ACTH stimulation was used for the diagnosis of adrenal insufficiency during acute brain injury. More sensitive than standard testing, its main advantage in mild HPA axis insufficiency is early diagnosis, because the higher pharmacological amount used in standard-dose ACTH testing may allow a partially dysfunctional gland to release a sufficient amount of hormone to appear falsely normal. However, low-dose ACTH testing is not routinely used or recommended in critically ill brain-injured patients because there is no diagnostic cut off value for intensive care unit settings. Low-dose ACTH testing may gain a place in the workup of HPA axis evaluation in critically ill patients and may be used routinely in the near future.
Because serum cortisol concentrations may vary, the magnitude of serum cortisol response to ACTH stimulation was also considered critical in the diagnosis of adrenal failure and the identification of a patient’s status as a responder (change from baseline cortisol of at least 9 µg/dl) or non-responder to stimulation. But a recent study showed no positive correlation between this criterion and survival. Current guidelines recommend this classification only in septic shock. Therefore, we do not recommend using this criterion in the diagnosis of adrenal insufficiency in patients with acute brain injury.
Finally, cortisol is measured in the blood as both an unbound (“free”) form, which is the biologically active hormone, and as cortisol bound with corticosteroid-binding globulin (CBG). CBG is commonly low in critical illness/injury, especially when serum albumin is <2.5 g/dl and it may be one of the causes of an apparent low cortisol serum concentration. Measurement of free cortisol has been suggested as a more accurate assessment of adrenal output. Testing methods for CBG and free cortisol are rarely available, however. Several research groups have recently investigated the use of salivary cortisol concentration as a surrogate marker for serum free cortisol levels. Salivary cortisol measurement is simple to obtain and easy to measure in most laboratories. The data on the utility of salivary cortisol in critically ill brain-injured patients are currently insufficient.
The diagnosis and the management of adrenal insufficiency are briefly summarized in Figure 23.2.
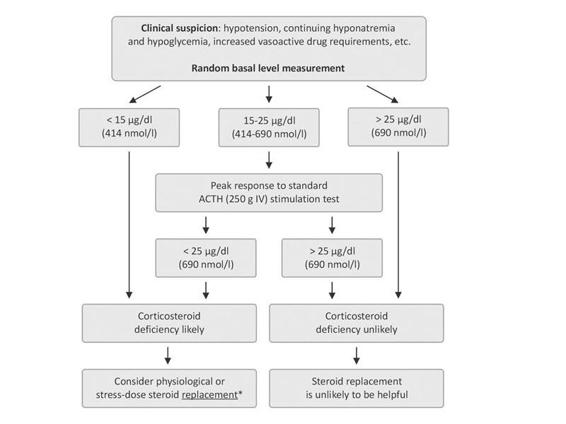
Figure 23.2. Algorithm for the management of adrenal insufficiency in brain-injured patients in the acute phase.
* It is proved that a pharmacological dose of corticosteroid replacement is not useful and may be harmful. In stable patients, physiological dose (hydrocortisone 10 mg IV every 8 h), in unstable patients with severe clinical findings, stress dose (hydrocortisone 50-100 mg IV every 8 h or an IV infusion of about 15 mg/h) steroid replacement is recommended.
Treatment
Large scale clinical studies such as the Corticosteroid Randomization After Significant Head Injury (CRASH) trial and literature reviews have indicated that routine corticosteroid administration at high doses or pharmacological doses (30 mg/kg methylprednisolone) to all patients with moderate/severe TBI is not indicated or may be harmful.
It is generally agreed, however, that therapy should be provided for patients with confirmed hypoadrenalism in whom clinical circumstances, such as hypotension, hyponatremia and hypoglycemia, warrant intervention. The steroid replacement dose should be titrated based on the critically ill patient’s clinical status and requirements. In patients with adrenal insufficiency as determined according to previously discussed criteria (Fig. 23.2) and stable, replacement with hydrocortisone (10 mg IV every 8 h) could be sufficient initially. When severe stress is present and the patient is not stable 50-100 mg of hydrocortisone IV every 8 h or an IV infusion of about 15 mg/h is recommended (Fig. 23.2). Dexamethasone or methylprednisolone at equivalent hydrocortisone doses could be used as alternatives. It is important to note that dexamethasone does not have any mineralocorticoid activity. Addition of a mineralocorticoid is not usually required, but may be considered when hyponatremia is significant and the patient has proven primary adrenal failure.
The optimal duration of corticosteroid supplementation is unknown, but it should be continued until the patient’s clinical situation has improved and when there is no need for vasopressor therapy.
The Thyroid Axis
Several changes in circulating thyroid hormone concentrations occur in acute critical illnesses including brain injury. The conversion of T4 (thyroxine) to T3 (triiodothyronine) in peripheral tissues is dramatically reduced and an altered form of T3, reverse T3 (rT3), is generated. Serum measurement will characteristically show a low T3; normal or low T4 and normal TSH have been synonymously referenced as the euthyroid-sick syndrome or low T3 and/or low T4 syndrome. These changes appear immediately after brain injury, can persist for at least 2 weeks and may normalize after the acute illness has resolved. True “central” hypothyroidism occurs in 4 to 15% of patients following brain injury. The presence of low free T4 and low TSH level is diagnostic for central or secondary hypothyroidism; except for academic purposes, stimulation testing for diagnosis is not warranted in clinical practice. Impaired thyroid axis, however, should be confirmed in the absence of other medications known to reduce TSH production/release (e.g., dopamine, high-dose glucocorticoids). Although the evidence is not strong, some studies demonstrated that central hypothyroidism was associated with prolonged coma following TBI, poor neurological outcome, and higher mortality as compared to patients in whom TSH did respond to TRH stimulation.
Thyroid hormone replacement during the acute treatment of TBI or nontraumatic brain injury has not been reported previously. Prospective studies demonstrated that TSH deficiency (central hypothyroidism) is generally transient and that most of the changes resolve after 3 months. In critically ill burn or medical patients, acute thyroid hormone replacement therapy was not shown to produce short-term improvement.
Therefore, based on current evidence we do not recommend thyroid hormone replacement in the acute phase of brain injury. If TSH deficiency persists for more than 2 weeks and the clinical findings are compatible with hypothyroidism, the patient could be treated after steroid replacement if adrenal insufficiency is present.
The Somatotropic Axis
A reduced GH response to GHRH or other stimulation soon after severe TBI occurs in approximately 18% of patients. Early after TBI, basal plasma GH is within normal limits or high, and insulin-like growth factor (IGF-I) is normal or low. Peripheral resistance to GH action (probably due to decreased GH-receptor expression), manifested by high basal GH concentrations with low IGF-I, has been reported in patients with critical illness including brain injury. This seems to be an adaptive response because GH is an anabolic hormone: in the acute phase of critical illness, catabolism is the initial response to provide fuel for vital organs. Based on basal hormone levels and dynamic tests, current studies (Table 23.1) demonstrate that 20 to 50% of brain-injured patients have GH deficiency. Prospective data confirm that most patients with GH deficiency during acute phase recover within 3 to 12 months after brain injury.
GH and IGF-I levels in the acute phase are not generally correlated with trauma severity. Some studies investigating the correlation between GH response to provocative tests and prognosis or clinical outcome in the early post-TBI period have reported conflicting results.
To date, no studies have investigated the effects of GH replacement therapy in the acute phase of brain injury in patients with GH deficiency. In a recent randomized, prospective, double blind, placebo controlled study by Hatton et al., IGF-I and GH therapy within 72 hours of TBI produced sustained improvement in metabolic and nutritional endpoints. But the GH status of the TBI patients in this study was not defined. A large multicenter study by Takala et al. showed that the administration of high doses of GH in critically ill patients doubled the mortality instead of improving the outcome.
Therefore, GH supplementation is not currently recommended during the acute phase of brain injury. Future confirmatory data are needed to determine the effects of physiological dose GH replacement in GH-deficient patients with prolonged critical illness at the catabolic state.
The Gonadotropic-lactotropic Axis
After brain injury, and after TBI in particular, a substantial decrease in testosterone and low gonadotropin (FSH/LH) levels develop shortly after injury and deteriorate over the ensuing days. The magnitude of the decline in testosterone was correlated with head trauma severity in some studies but not in others. The pituitary-ovarian axis is comparably depressed.
The incidence of hypogonadism in critically ill TBI patients varies between 20 and 80%. In most patients, hypogonadism improves within 3-6 months after injury. The current data suggest that these changes in the gonadotropic axis in the acute phase of brain injury are adaptive and transient; therefore, gonadotropin replacement is not recommended in the acute phase.
Prolactin (PRL) may increase, remain normal, or decrease in acute brain injury. Baseline PRL is negatively correlated with head trauma severity, as expressed by the Glasgow Coma Scale score. Hyperprolactinemia is reported in 20 to 50% of patients with acute brain injury. Although the mechanism and the effects are not known, this response is thought to be adaptive.
23.1.6 Posterior Pituitary Dysfunction in the Acute Phase of Brain Injury
Inadequate ADH secretion due to posterior pituitary dysfunction results in central diabetes insipidus (DI). DI following TBI is less common than anterior lobe injury, occurring acutely in around 20-25% of patients. Most patients recover from DI, but rarely (6-7%) persistent DI may occur. While DI is a well-recognized complication of TBI, the data one patients with other causes of brain injury are scarce.
The primary symptoms of DI are thirst and polyuria in conscious patients. In critically ill patients, the main clinical findings are increased daily urinary volume (>3 litres) and hypernatremia. If volume is significantly reduced, hypernatremia may cause weakness, altered mental status, seizures and coma. The diagnosis of DI in brain-injured patients with clinical suspicion is confirmed by measuring plasma and urine osmolality. The urine is inappropriately diluted for the degree of serum osmolality, usually urine osmolality < 200 mOsm/kg and specific gravity < 1005 with plasma osmolalite >295 mOsm/kg. The most commonly used agent to treat central DI is the ADH analogue desmopressin, which has a longer half-life and almost no pressor activity. It may be administered intranasally at a dose of 5-20 µg once or twice daily. In critically ill patients, desmopressin is more commonly given IV, IM or subcutaneously (2-4 µg once or twice daily), which is 5-10 times more potent than via intranasal route. The effectiveness of the treatment is established by measuring urinary volume and plasma sodium concentration.
Despite the hypernatremic dehydration induced by ADH deficiency, hyponatremia also develops in many patients during the acute period of brain injury due to an inappropriate release of ADH (syndrome of inappropriate antidiuretic hormone, [SIADH]). The reported prevalence of this abnormality is 2.3-36%. As a rule, TBI-induced SIADH is transient.
Symptoms of SIADH vary depending on the degree of hyponatremia (serum sodium levels < 130 mEq/l and < 125 mEq/l in severe cases) and rapidity at which it develops. SIADH is a diagnosis of exclusion of other hyponatremia causes. Renal, adrenal and thyroid function must be normal to establish the diagnosis. In BI patients with HPA axis dysfunction, SIADH can only be diagnosed after appropriate corticosteroid replacement. A low (280 mOsm/kg) serum osmolality is consistent with SIADH. An inappropriately elevated urine osmolality (>100 mOsm/kg) and urine sodium (>40 mEq/l) concentration are consistent with SIADH. Most cases are self-limiting and the cornerstone treatment is fluid restriction. Early diagnosis and treatment of posterior pituitary dysfunction after brain injury is important for maintaining water homeostasis and electrolyte balance in critically ill patients.
23.1.7 Pathophysiology of Pituitary Dysfunction Following Acute Brain Injury and Predictive Value of Hormonal Changes
There are several mechanisms underlying hypothalamo-pituitary dysfunction due to TBI including hypoxic insult or direct mechanical injury to the hypothalamus, the pituitary stalk, or the pituitary gland; compression from hemorrhage, edema, or increased intracranial pressure; and vascular injury to the hypothalamus or the pituitary gland. Autopsy findings in patients who died of TBI showed evidence of injury to the hypothalamus, the pituitary gland or the pituitary stalk in 26-86% of cases. Rotational acceleration-deceleration can cause shearing injury of axons, as commonly seen in the midline structures of the brain. This may represent a possible mechanism of hypothalamic pituitary dysfunction after TBI. In nontraumatic brain injury, evidence for the possible mechanisms of pituitary dysfunction is lacking; however, vascular injury seems to be the most possible cause in the pathogenesis of nontraumatic brain injury. The exact mechanisms of acute hormonal response and pituitary dysfunction due to brain injury remain to be elucidated and further clinical and experimental studies are warranted.
Recent prospective studies in patients with TBI and SAH revealed that most of the acute phase hormonal changes are transient and do not predict pituitary dysfunction at 6 months and 12 months after brain injury. Current guideline recommendations advise evaluation of pituitary function in all TBI patients with moderate and severe injury at 3 to 6 months and again at 12 months after trauma, regardless of acute hormone deficiencies.
23.1.8 Conclusions and Key Concepts
Related posts:
 Acute Renal Injury in the Neurocritical Patient
Neurocritical Care of Patients With Arteriovenous Malformations
The Diagnosis and Management of Central Nervous System Infections in the Neurocritical Care Unit
Stroke Units: Organization, Past, Present and Future
Gastrointestinal Disorders in the Neurocritical Patient
Concepts and Management of Brain Death and Management of Potential Organ Donation
Acute Renal Injury in the Neurocritical Patient
Neurocritical Care of Patients With Arteriovenous Malformations
The Diagnosis and Management of Central Nervous System Infections in the Neurocritical Care Unit
Stroke Units: Organization, Past, Present and Future
Gastrointestinal Disorders in the Neurocritical Patient
Concepts and Management of Brain Death and Management of Potential Organ Donation
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


