Epidemiologic Aspects of Epilepsy
Anne T. Berg
Until the 1960s, the prevailing view about epilepsy was captured by the words of Sir William Gowers: “The tendency of the disease is toward self-perpetuation; each attack facilitates the occurrence of another by increasing the instability of the nerve elements.… The spontaneous cessation of the disease is an event too rare to be reasonably anticipated” (1). Epilepsy was considered a progressive, nonremitting disease. Gowers, who studied and characterized patients seen in the early tertiary referral centers of the late 1800s, was extremely observant, however. In the same text, he noted that “… the cases in which the best results are obtained, in which no more fits occur, are precisely those who are never heard of again. Such cases are, assuredly, far more numerous than imagined. The few which are incidentally heard of at a subsequent time make it quite certain that they represent a much larger number of whom no trace is obtained.” In this brief comment, Gowers identified what would help pave the way for the important role played by epidemiology in the study of seizures: to obtain those traces and to study not only those patients in need of continuous care, but those whose seizures resolved as well.
Early epidemiologic studies provided the basis for at least two major contributions to the understanding of epilepsy and seizures in the general population. These studies demonstrated that seizures are relatively common. The cumulative lifetime risk of the occurrence of any type seizure may reach as high as 10%, possibly higher (2,3). These early studies also provided a better basis for understanding the prognosis of epilepsy and for distinguishing the disorder from other conditions in which seizures might occur. In short, these studies showed that the majority of individuals with epilepsy fared very well (4). Contrary to Gowers’ characterization (although not inconsistent with his subsequent comment), the majority of patients became completely seizure-free for extended periods and often for the rest of their lives. The early epidemiologic studies also provided some impetus and data for distinguishing situational-related seizures and single seizures from epilepsy proper (5). This led to substantial changes in clinical practice, in counseling, and, most likely, in the lives of individuals affected by seizures.
Since these studies in the 1960s and 1970s, epidemiologic investigations have become more sophisticated and more informative, having incorporated new knowledge and distinctions about seizure disorders. This chapter discusses the current definitions and distinctions used in epidemiologic research, as well as some of the more recent and challenging aspects of epidemiologic studies of seizure disorders, especially as they affect comparative work and investigations in developing countries.
CURRENT DEFINITIONS AND DISTINCTIONS USED IN EPIDEMIOLOGIC EPILEPSY RESEARCH
Epilepsy (recurrent, unprovoked seizures) is now distinguished from many other conditions and situations in which seizures may occur. The following accepted definitions are in current use and are used throughout this chapter.
Epileptic Seizure
An epileptic seizure is characterized by the clinical manifestation of a discrete, abnormal discharge of neurons in a part of the brain or throughout the brain. Epileptic seizures must be distinguished from nonepileptic seizures and from other conditions that can produce clinical manifestations that are highly similar to those caused by epileptic seizures (6).
Acute Provoked Seizure
An acute provoked seizure is one that occurs in the context of an acute brain insult or systemic disorder, such as, but not limited to, stroke, head trauma, a toxic or metabolic insult, or an intracranial infection (7).
Unprovoked Seizure
A seizure that occurs in the absence of an acute provoking event is considered unprovoked (7).
Epilepsy
Epilepsy is generally diagnosed after the occurrence of at least two unprovoked seizures on separate days, at least 24 hours apart. An individual with a single unprovoked seizure or with two or more unprovoked seizures within a 24-hour period is not considered to have epilepsy per se (7), although it is still entirely reasonable to identify the underlying form of the epileptic disorder at the time of the initial seizure (8).
Etiology
Etiology is considered symptomatic if there is an antecedent condition that is causally related to an increased risk of developing epilepsy. Symptomatic factors include, but are not limited to, history of stroke, brain malformation, clear neurodevelopmental abnormality such as cerebral palsy, history of bacterial meningitis or viral encephalitis, a variety of genetic conditions, and tumors. Epilepsy in the context of a progressive condition (e.g., neurodegenerative disease or an aggressive tumor) is often considered a subgroup within symptomatic epilepsy. In contrast, epilepsy is considered nonsymptomatic in the absence of such antecedent factors. The nonsymptomatic epilepsies are further divided into idiopathic (i.e., those in which other clinical evidence including electroencephalograms [EEGs] indicates that the individual has a form of idiopathic epilepsy) and cryptogenic (in which the underlying cause remains unclear). Recently, the distinction between these two designations has become blurred by the realization that there may be some commonalities between syndromes traditionally considered idiopathic and others considered cryptogenic (9,10).
Gray Areas
Neonatal seizures (i.e., those occurring in an infant <28 days old) are usually differentiated from epilepsy for a variety of reasons; however, several specific forms of epilepsy have been reported in this age group (11). Febrile seizures are a well-described and recognized seizure disorder that, for historical reasons, has been distinguished (both clinically and in research) from epilepsy. This has had important clinical implications for the treatment of children with such seizures. From a scientific standpoint, however, this may obscure significant aspects of the genetics and underlying neurophysiologic mechanisms of seizure-related disorders.
Epilepsy Syndromes
Epilepsy syndromes are presented in greater detail in subsequent chapters of this book; however, they are important to modern epidemiologic endeavors and thus merit mention in this context. As with cancer, epilepsy is not a single disease. In theory, epilepsy syndromes represent forms of epilepsy that have different causes, different manifestations, different implications for short- and long-term management and treatment, and different outcomes. The classification of epileptic syndromes was first proposed more than 30 years ago. The current version, which was published in 1989, is undergoing major restructuring and revision (12, 13, 14, 15, 16, 17).
EPIDEMIOLOGY
As already mentioned, early epidemiologic studies were key to understanding the frequency of seizures in the population and to providing the initial impetus for some of the distinctions previously outlined. Prior to the advent of epidemiology, one could say that “the forest had been lost for the trees”—and the most intractable trees at that! Epidemiology provided the context, the forest, and a bit about what was in that forest. Many of the distinctions described above cannot be made without considering diagnostic technologies and capabilities.
Diagnostic Technology and Expertise
Seizures and epilepsy present a complex situation because the diagnosis is not based on a single source or type of information. Rather, epilepsy is a clinical diagnosis supported to a greater or lesser extent by a wide range of data obtained from several sources: the medical history, the history (both from the patient and witnesses) of the events believed to be seizures, a neurologic examination, a reliable EEG, and sometimes neuroimaging (18). To have a valid diagnosis, one must also be able to rule out many other conditions that mimic seizures. These disorders include, but are not limited to, movement disorders, parasomnias, attention deficit hyperactivity disorder (ADHD), pseudo- or nonepileptic seizures, transient ischemic attacks, and syncope. In addition to the simple diagnosis of epilepsy, adequate information is needed to identify as accurately as possible the specific syndrome and its underlying cause. In turn, this depends largely on clinical features and on the EEG, and often on neuroimaging.
Screening questionnaires have been used in epidemiologic studies for the purpose of obtaining a rough estimate of the frequency of seizure disorders (19, 20, 21). Their diagnostic
specificity, however, can be quite poor (i.e., they identify individuals as having epilepsy who, in fact, do not have the disorder). Individuals identified via such an approach are then evaluated more thoroughly by physicians. Unfortunately, the training of these physicians or other health care professionals and the access to such basic diagnostic tools as EEGs are often less than ideal.
specificity, however, can be quite poor (i.e., they identify individuals as having epilepsy who, in fact, do not have the disorder). Individuals identified via such an approach are then evaluated more thoroughly by physicians. Unfortunately, the training of these physicians or other health care professionals and the access to such basic diagnostic tools as EEGs are often less than ideal.
The importance of professional training has been highlighted in several studies. The British National General Practice Study of Epilepsy (NGPSE) included all individuals referred by general practitioners. Of these patients, only 71% were confirmed by an expert as having epilepsy (22). The Dutch study of epilepsy in childhood reported that greater than 25% of children referred for epilepsy consultation by general practitioners and pediatricians did not have the disorder (23). By contrast, the epidemiologic study of incident epilepsy in the Gironde region of France (EPIGIR), which identified patients through hospital neurology services and EEG laboratories, excluded only about 10% of patients (24). In the Connecticut study, only about 2% of referrals from child neurologists were not confirmed (by the research team) as having epilepsy (25). Finally, the Coordination Active du Réseau Observatoire Longitudinal de L’Epilepsie (CAROLE) study, which also relied on referral after diagnosis by a network of neurologists, did not exclude any patients because of an unconfirmed diagnosis (8). In all these studies, epileptologists reviewed the medical data; however, the level of expertise of the individual(s) who collected the initial information and how that information was interpreted and recorded varied.
Health care systems vary enormously in terms of (a) sophistication of care, (b) access to that care, and (c) use of available care. These differences affect how the same study might be conducted in different settings. To obtain adequate evaluations and diagnostic accuracy, either the study itself must provide all of the evaluations, or the evaluations must depend on the health care system. The first approach can be prohibitively expensive, and the feasibility of the second approach depends on where the study is conducted. In health care systems that rely heavily on specialist care, new-onset seizures are often evaluated by a specialist (26, 27, 28, 29). In settings that rely on general practitioners, on the other hand, only the more difficult cases tend to be seen by a specialist with the necessary expertise to render an accurate diagnosis (30). In many developing nations, there is no system in place for epilepsy care and for most other medical needs (31, 32, 33, 34, 35).
Considerations in Ascertaining Cases
Epidemiologic studies often focus on a disorder as it occurs in a defined population, with the goal of determining the incidence or prevalence of the disorder and of its subtypes. This requires that a complete ascertainment method be used or that the sampling fraction be known. Even when complete ascertainment or systematic partial sampling is not feasible, a sample that is representative of the patients in the general population from which it was drawn allows one to make inferences regarding the relative significance of various causes and the frequency of particular types of epilepsy. There are several biases to avoid.
Prevalence Bias
Prevalent cases refer to individuals who, at a given time, carry a diagnosis of epilepsy or are under the care of a health care professional for epilepsy. In a disorder with a relatively low mortality rate, prevalent cases are overrepresented compared with those who remit courses (Fig. 8.1) (36). Gowers’ comment is a prime example of an erroneous conclusion based on prevalent samples. As another example, Viani and colleagues reported the distribution of syndromes in a newly diagnosed versus a prevalent sample (37). Benign rolandic epilepsy accounted for 8.3% of newly diagnosed cases but only 2.3% of prevalent cases. Lennox-Gastaut syndrome accounted for 5% of newly diagnosed cases but 17% of prevalent cases. When mortality is high, prevalent samples tend to underestimate the number of severe cases (who die early) in favor of survivors. Although prevalent samples provide some relevant information for health care services, for the purposes of understanding the occurrence, causes, and outcomes of a disorder, they are methodologically inappropriate and prone to bias because they selectively emphasize the ongoing cases of a disease. Incident cases are generally preferred.
Ascertainment Bias
Poor response rate, inadequate ascertainment methods, and other problems that decrease the proportion of eligible cases recruited into a particular study can all affect the generalizability of the results of that study. This is because
those who do not participate or who are not included are often systematically different from those who are included.
those who do not participate or who are not included are often systematically different from those who are included.
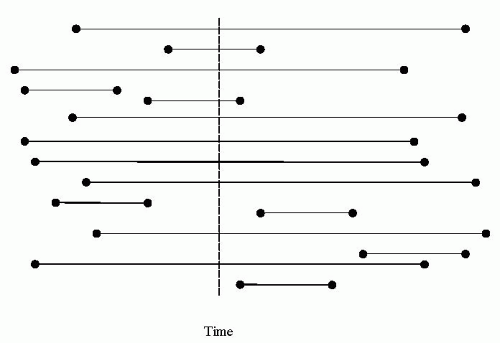 Figure 8.1 Prevalence bias. Each horizontal line represents a case with active disease (i.e., a prevalent case). The length of the line represents the time of the active disease, with onset to the left and offset or death to the right. The dashed vertical line represents the day on which prevalence is measured. Long-duration cases are oversampled (eight of eight are ascertained on the prevalence day) relative to short-duration cases (two of seven are ascertained on the prevalence day). |
Referral Center Bias
Recruitment through a specialty center biases a study group toward more extreme, difficult-to-diagnose and difficult-to-treat cases. This was demonstrated with febrile seizures in relation to the risk of epilepsy (38). Children with febrile seizures studied in population-based or near population-based cohorts had a uniformly low risk of subsequent epilepsy. In contrast, the estimated risks of epilepsy in studies that recruited children from special referral centers were very inconsistent, with many exceeding 30%.
To avoid these and related biases requires an understanding of the typical process by which persons with epilepsy are diagnosed and a concerted effort to identify and recruit all potentially eligible individuals in a population during a specified time frame. In developed nations, these issues may seem relatively trivial. In developing nations, however, they can pose tremendous challenges to the researcher. Pal and associates eloquently describe their experiences and observations from studies they conducted on childhood epilepsy in rural India (39, 40, 41, 42). The paucity of health care services, the expenses and difficulties involved in using those services, the mistrust of westernized medicine, local beliefs about healers, and the stigma associated with epilepsy were just some of the factors that had to be addressed in order to proceed with their research.
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


