Epilepsy in the Setting of Neurocutaneous Syndromes
Prakash Kotagal
Deepak K. Lachhwani
The neurocutaneous syndromes are a group of unrelated disorders characterized by congenital dysplastic abnormalities involving the skin and nervous system. Epileptic seizures are encountered most frequently in tuberous sclerosis and the Sturge-Weber syndrome. Neurofibromatosis and other lesser-known entities, such as epidermal nevus syndrome, are also known to be accompanied by epilepsy.
TUBEROUS SCLEROSIS
Genetics
Tuberous sclerosis, also known as Bourneville disease, is a congenital hamartomatosis that affects multiple organ systems, especially the central nervous system (CNS). A genetic disorder with high penetrance and variability in expression, it is inherited as an autosomal dominant condition; the prevalence of the gene is estimated at 1 in 9700 persons (1). The gene locus in one-third of tuberous sclerosis families has been mapped to chromosome 9q34 (also known as TSC1, or tuberous sclerosis complex-1) (2); in other families, the gene locus is on 16p13 (TSC2) (3). Linkage studies initially indicated that there was a high incidence of apparently sporadic cases caused by new mutations; these appeared to account for 60% of all new cases (4, 5, 6). However, data using single-strand conformational polymorphism and heteroduplex analysis showed spontaneous mutations to account for only 22% of all cases (7). Hamartin, the product of the TSC1 gene, appears to be localized to cytoplasmic vesicles. Tuberin, another protein encoded by the TSC2 gene, functions as a guanosine triphosphatase (GTPase)-accelerating protein for the smallmolecular-weight GTPases Rap1a and Rab5. Tuberin has been colocalized to the perinuclear region of cultured cells and has been shown to colocalize with Rap1 in the stacks of the Golgi apparatus. A coiled-coil domain near the COOH terminus of hamartin was shown to bind specifically to a coiled-coil domain near the NH2 terminus of tuberin, indicating that the two proteins work together in the same cellular pathway (8). It has been hypothesized that tumorigenesis in tuberous sclerosis could be the result of aberrant vesicular trafficking (9). Lack of tuberin has been shown to trigger inappropriate proliferation and differentiation in the neuroepithelium of the telencephalic vesicles during midgestation (10). Evidence suggests that TSC genes participate in multiple signaling cascades, one of which is binding to mTOR (mammalian target of rapamycin), an enzyme that is critical to normal cell growth and proliferation. Disruption a result of loss of TSC1 and TSC2 function activates the mTOR cascade, resulting in abnormal growth and proliferation. An understanding of this mechanism provides a unique opportunity to test rapamycin (a highly specific inhibitor and previously approved by the Food and Drug Administration as therapy for some renal neoplasms) as a potential therapeutic agent (11).
Tuberin has also been identified in cortical dysplasia tissue, suggesting that, in addition to its role as a growth suppressor, it may also contribute to cortical maturation and cellular differentiation (12,13).
Both parents of a child who has tuberous sclerosis should be examined for evidence of the disease by means of a complete physical examination, examination under a Wood lamp, dilated eye examination, magnetic resonance imaging (MRI) of the head, and renal ultrasonography. If the parents
have no evidence of tuberous sclerosis, the recurrence risk for their next child is approximately 1:10,000 (the same as in the general population); if either parent has the disease, the risk for another affected child is 50%.
have no evidence of tuberous sclerosis, the recurrence risk for their next child is approximately 1:10,000 (the same as in the general population); if either parent has the disease, the risk for another affected child is 50%.
Clinical Features
The classic Vogt triad of seizures, mental retardation, and facial angiofibroma is seen in only 29% of patients with tuberous sclerosis (14,15). Gomez (14,15) found that 6% of tuberous sclerosis patients had none of the symptoms of the triad. One of the following major criteria must be present to establish a diagnosis of tuberous sclerosis: facial angiofibroma or periungual fibroma; cortical tuber, subependymal nodule, or giant cell astrocytoma; multiple retinal hamartomas; or multiple renal angiomyolipomas. The diagnosis can also be made if the patient has two of the following minor criteria: infantile spasms; hypomelanotic macules (ash leaf spots); single retinal hamartoma; subependymal or cortical calcification on a computed tomography (CT) scan; bilateral renal angiomyolipomas or cysts; cardiac rhabdomyoma; or history of tuberous sclerosis in a first-degree relative (14,15). More than 90% of patients have calcifications in the subependymal region of the lateral ventricle or cortical tubers. In Gomez’s series of 300 tuberous sclerosis patients, skin lesions were seen in 96%, seizures in 84%, retinal hamartomas in 47%, and mental impairment in 45% (15).
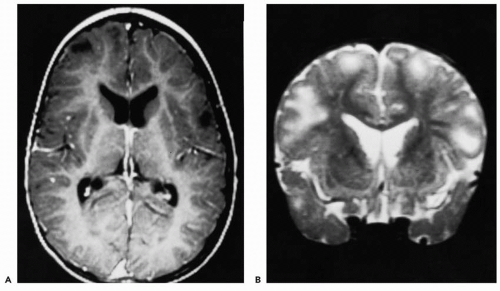 Figure 36.1 Magnetic resonance scan of a 6-year-old boy with tuberous sclerosis shows multiple cortical and subcortical tubers. These appear as hypointense areas on T1-weighted images (A) and as areas of increased signal on T2-weighted images (B). Also evident in B are subependymal nodules protruding into the lateral ventricles. |
Cortical and subcortical tubers are hamartias and constitute a hallmark of the disease. Cortical tuber count and distribution account for the phenotypic variability, with a higher tuber burden being associated with more severe clinical manifestations (16). Tubers also occur in the cerebellum in 15% of patients (17). Ventricular dilation is a common but nonspecific finding. Subependymal nodules are hamartomas, typically located in the lateral ventricles near the foramina of Monro. They are found in a majority of patients and are usually asymptomatic unless their growth results in blocking of the cerebrospinal fluid circulation. The development of brain tumors in these patients is uncommon; however, subependymal giant cell astrocytomas may occur in up to 8% of patients, typically near the foramen of Monro. They are believed to originate from a subependymal nodule (18). Patients may also have heterotopic masses of gray matter, consistent with the concept that tuberous sclerosis is a dysplastic process. Roach and colleagues (16) found that patients with five or more cortical lesions were more likely to have intractable seizures and developmental delay.
MRI has become the imaging tool of choice in the past two decades. Although CT scanning is more fruitful in demonstrating calcified lesions, MRI is superior in detecting these and other smaller lesions, which are likely to be missed on CT scans. In the myelinated brain, tubers appear as regions of low T1 and high T2 signal, and may show an enlargement of the corresponding gyri (Fig. 36.1). Fast fluid
attenuated inversion recovery (FLAIR) sequences are more sensitive in picking up smaller lesions that appear as hyperintense areas (19). During early infancy, the appearance of tubers may vary, demonstrating a paradoxically increased T1 signal and a decreased T2 signal. The reason for this paradox is unclear and may be related to the immature white matter (20). The role of FLAIR sequences in the developing brain is yet to be determined. Subependymal nodules are better seen on T1-weighted sequences, whereas cortical tubers are better seen with T2-weighted sequences (21). On T1-weighted images, subependymal nodules are isointense to white matter and slightly hyperintense compared with gray matter; they are isointense or hypointense on T2-weighted images, which may show a central area of decreased signal. Tubers are further classified as sulcal islands (i.e., ring of hyperintensity surrounding an island of isointense cortex, seen on T2-weighted images) and gyral cores in which the white matter of a gyrus is hypointense, surrounded by isointense normal-appearing cortex on T1-weighted images (17). Sometimes multiple cortical tubers may give the appearance of pachygyria (22). Adjunctive imaging techniques such as magnetic resonance (MR) spectroscopy, diffusion-weighted imaging, position emission tomography (PET) scanning (with ligands such as fluoro-2-deoxyglucose, [11C]-α-methyl-L-tryptophan, flumazenil, and [18F]trans-4-fluoro-N-2-[4-(2-methoxyphenyl) piperazin-1-yl]ethyl]-N-(2-pyridyl) cyclohexanecarboxamide [FCWAY]), and single-photon-emission computed tomography (SPECT) scanning are newer tools used in evaluation of these patients (23,24). Their role is constantly evolving and will be clearer with additional data and improvement in techniques (25, 26, 27).
attenuated inversion recovery (FLAIR) sequences are more sensitive in picking up smaller lesions that appear as hyperintense areas (19). During early infancy, the appearance of tubers may vary, demonstrating a paradoxically increased T1 signal and a decreased T2 signal. The reason for this paradox is unclear and may be related to the immature white matter (20). The role of FLAIR sequences in the developing brain is yet to be determined. Subependymal nodules are better seen on T1-weighted sequences, whereas cortical tubers are better seen with T2-weighted sequences (21). On T1-weighted images, subependymal nodules are isointense to white matter and slightly hyperintense compared with gray matter; they are isointense or hypointense on T2-weighted images, which may show a central area of decreased signal. Tubers are further classified as sulcal islands (i.e., ring of hyperintensity surrounding an island of isointense cortex, seen on T2-weighted images) and gyral cores in which the white matter of a gyrus is hypointense, surrounded by isointense normal-appearing cortex on T1-weighted images (17). Sometimes multiple cortical tubers may give the appearance of pachygyria (22). Adjunctive imaging techniques such as magnetic resonance (MR) spectroscopy, diffusion-weighted imaging, position emission tomography (PET) scanning (with ligands such as fluoro-2-deoxyglucose, [11C]-α-methyl-L-tryptophan, flumazenil, and [18F]trans-4-fluoro-N-2-[4-(2-methoxyphenyl) piperazin-1-yl]ethyl]-N-(2-pyridyl) cyclohexanecarboxamide [FCWAY]), and single-photon-emission computed tomography (SPECT) scanning are newer tools used in evaluation of these patients (23,24). Their role is constantly evolving and will be clearer with additional data and improvement in techniques (25, 26, 27).
Epilepsy
The prevalence of epilepsy in patients with tuberous sclerosis has been reported in several series to be higher than 80% (14,15,28, 29, 30, 31, 32). These studies also found mental retardation in more than 60% of children, especially when seizures started before the age of 2 years. In a prospective study of families with tuberous sclerosis, however, Webb and coworkers (33) found a frequency of 62% for epilepsy and 38% for mental retardation. Seizures began before the age of 2 years in 71% of Westmoreland’s cases (34). Infantile spasms are the most common type of seizure at presentation (36% to 69% of patients) (31,34, 35, 36), followed by focal motor (24%), grand mal (27%), and, less often, complex partial or atypical absence seizures (34). Tuberous sclerosis accounts for 25% of patients presenting with infantile spasms (37). In rare instances, patients with tuberous sclerosis may present with seizures during the neonatal period (38). Yamamoto and associates (36) found that 70% of children with tuberous sclerosis and seizures developed complex partial seizures after a follow-up period of 5 to 20 years. When seizures start after the age of 2 years, children usually have complex partial or secondarily generalized seizures (39). Roger and associates (40) found that in patients with seizure onset after 12 months, partial seizures remained as the sole type, with one third of these children showing a favorable evolution.
The results of electroencephalography EEGare abnormal in most tuberous sclerosis patients. Westmoreland found that 88% of patients had abnormal recordings, most frequently epileptiform discharges (75%); slowing occurred in 13%. The epileptiform discharges were multifocal in 25% and focal in 23%; hypsarrhythmia occurred in 19% and generalized spike-wave discharges in 8%. Approximately 70% of focal spikes were in the temporal lobe (34). Background asymmetries have been reported in 40% of children with tuberous sclerosis, especially those with poorly controlled epilepsy (36). Cusmai and coworkers (41) reported that during follow-up, although some epileptic foci disappeared (usually occipital foci), others became evident, especially in the frontal regions (consistent with posteroanterior migration of epileptic foci in childhood [42]). Secondary bilateral synchrony appeared in 35% of children with tuberous sclerosis after the age of 2 years, especially during drowsiness and sleep (42,43). This agrees with our observations of children with multifocal spike discharges (44). Secondary bilateral synchrony was related to the presence of frontal lobe tubers. Cusmai and coworkers (41) also found that, although there was no correlation between the number of EEG foci and the number of cortical tubers, in 25 of 26 patients, there was an electroencephalographic (EEG) and MRI topographic correlation between at least one large tuber (larger than 10 mm in the axial plane and larger than 30 mm in the coronal plane) and one EEG focus. Similar results were reported by Tamaki and associates (45), who also found a higher incidence of hypsarrhythmia in patients with bilateral tubers.
Mental retardation occurs in 38% to 65% of children with tuberous sclerosis (14,15,33,40,46); approximately one third each being normal or borderline, mildly retarded, or profoundly retarded (46). They may also have dyspraxia, speech delay, visuospatial disturbance, dyscalculia, or memory problems. Marked hyperkinesia and aggressive behavior are also seen (46,47). Hunt found that the incidence of aggressive behavior was 13% and that these patients did not have a history of infantile spasms. Between 26% and 58% of children with tuberous sclerosis and infantile spasms have infantile autism (46,47), compared with 13% of patients with infantile spasms who do not have tuberous sclerosis (48). Both the number of tubers and their topography seem to play an important role in mental outcome. The persistence of epileptic foci in anterior and posterior areas is thought to be important in the development of autistic traits, such as severe disability in verbal and nonverbal communication, stereotypes, and complete indifference to social interaction (46). Patients with multiple cortical lesions are likely to have developmental delay and intractable seizures (16). The association between infantile autism and tuberous sclerosis therefore appears to be more than coincidental.
Medical management of the seizures depends on the seizure types encountered. The response of children with tuberous sclerosis and infantile spasms to corticotropin is similar to the response of children with cryptogenic infantile spasms; however, those with tuberous sclerosis have a higher relapse rate (48). Riikonen and Simell (48) suggested that these children may benefit from an extended course of corticotropin. Other generalized seizures (i.e., myoclonic, absence, and tonic-clonic) are treated with benzodiazepines (i.e., clonazepam, nitrazepam, or clorazepate) alone or in combination with valproic acid; intractable seizures may be managed with corticotropin or a ketogenic diet (1). Complex partial and focal motor seizures respond best to carbamazepine, phenytoin, or primidone. Vigabatrin is thought to be particularly effective for partial-onset and generalized seizures (i.e., infantile spasms and tonic seizures of the Lennox-Gastaut syndrome) in patients who have tuberous sclerosis (49, 50, 51, 52). However, its ocular toxicity makes it unsuitable for long-term use.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


